夏世梅,袁梅,冯星星,蒋鸿超,段绍琴,李维熙,奎莉越,周百灵
11例X连锁无丙种球蛋白血症患儿的临床分析
夏世梅1,袁梅1,冯星星1,蒋鸿超2,段绍琴3,李维熙4,奎莉越1,周百灵1
1.昆明市儿童医院检验科,云南昆明 650100;2.昆明市儿童医院感染科,云南昆明 650100;3.昆明市儿童医院血液肿瘤科,云南昆明 650100;4.云南中医药大学中药学院,云南昆明 650500
分析云南地区X连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)患儿的临床特点及Bruton酪氨酸激酶(Bruton’s tyrosine kinase,BTK)基因的突变情况。回顾性分析2018年1月至2022年6月昆明市儿童医院经基因检测确诊的11例XLA患儿的临床资料,采用Sanger测序方法分析BTK基因的突变情况。11例患儿均为男性,发病年龄0.25~6.00岁,中位年龄1.50岁;基因诊断确诊年龄0.33~12.00岁,中位年龄4.17岁。临床表现以呼吸道感染为主,大部分患儿感染较重,其次为生长发育迟缓。患儿免疫球蛋白水平均较低,外周血CD19+B淋巴细胞百分比几乎缺如。基因检测结果显示剪接突变4例,错义突变3例,无义突变2例,缺失突变2例。其中发现1例新发突变,突变位点为c.1908+1G﹥C。对婴幼儿时期发生严重感染及生长缓慢的男童,应尽早行基因检测明确诊断。
X连锁无丙种球蛋白血症;Bruton酪氨酸激酶;原发性免疫缺陷
X连锁无丙种球蛋白血症(X-linked agamma- globulinemia,XLA)是一种儿童原发性免疫缺陷病,1952年Bruton最早报道该病,它是由Bruton酪氨酸激酶(Bruton’s tyrosine kinase,BTK)基因突变而使B淋巴细胞不能由前B淋巴细胞分化成熟进入外周血,最终导致B淋巴细胞缺乏。由于血液和组织中不能产生浆细胞,因此各种免疫球蛋白(immunoglobulin,Ig)生成严重减少,抗体反应明显缺乏[1-2]。XLA患儿以出生后6个月甚至更早月龄反复出现细菌感染为特征,其中呼吸道感染尤为突出。利用二代测序技术进行BTK基因突变检测是XLA的确诊指标,而定期静脉注射免疫球蛋白(intravenous immunoglobulin,IVIG)进行终身替代治疗可预防及治疗患儿感染,挽救患儿生命。现回顾性分析昆明市儿童医院近4年来确诊的XLA患儿的临床资料及基因检测结果。
1 资料与方法
收集2018年1月至2022年6月昆明市儿童医院经基因诊断确诊的11例XLA患儿的临床资料。临床诊断XLA采用1999年泛美免疫缺陷病组和欧洲免疫缺陷病协会制定的标准[3-4]。明确诊断标准为:男性患儿CD19+B淋巴细胞<2%,并符合以下至少1项:①BTK基因突变;②检测中性粒细胞或单核细胞发现缺乏BTK mRNA;③单核细胞或血小板缺乏BTK蛋白;④母系的表兄、舅舅或侄子CD19+B淋巴细胞<2%。征得患儿家属知情同意,11例患儿及父母皆采集外周血进行全外显子基因测序。本研究经昆明市儿童医院伦理委员会批准(2022-03-299-K01)。
2 结果
2.1 人口数据统计
11例男性患儿来自10个不同家庭,其中2例为彝族(例6和例8),其余为汉族。患儿均来自云南省各个地区,发病年龄0.25~6.00岁,中位年龄1.50岁,其中年龄最小的3个月;基因诊断确诊年龄0.33~12.00岁,中位年龄4.17岁。11例患儿中4例有家族史,例3和例4系同胞兄弟。
2.2 血清Ig水平和外周血CD19+B淋巴细胞占比
患儿免疫学评估均在未输注IVIG前进行。血清IgG均值为0.54g/L,11例患儿均<2g/L,其中2例IgG水平较高的患儿确诊年龄在3~6岁。血清IgM均值为0.2g/L,其中<0.2g/L的有7例,占63.6%。血清IgA均值为0.11g/L,<0.2g/L的有9例,占81.8%。11例患儿的外周血CD19+B淋巴细胞百分比几乎缺如,<2%的占100%。其中年龄最小的患儿(例10)IgG水平为0.93g/L,CD19+B淋巴细胞百分比为0.03%,见表1。
2.3 相关感染和伴随症状
患儿最常见的是呼吸道感染(10例),其中伴支气管肺炎5例,支气管扩张2例,肺实变4例,肺泡蛋白沉积症及气胸1例。部分患儿发生严重感染,中枢神经系统感染3例,败血症1例,脓毒血症2例,骨髓炎1例。其余情况包括生长发育落后3例,腹泻2例,营养不良2例,先天性心脏病2例,电解质紊乱2例,贫血2例,鹅口疮、麻疹、鼻窦炎、化脓性中耳炎及关节炎各1例。
病原微生物学检查:痰培养肺炎链球菌阳性3例,血培养肺炎链球菌阳性2例,脑脊液、骨髓、血培养木糖氧化无色杆菌反硝化亚种1例(例2),腿上伤口分泌物培养金黄色葡萄球菌阳性1例(例5),尿液培养大肠埃希菌阳性同时痰液检出流感嗜血杆菌、百日咳杆菌阳性1例(例3),肺泡灌洗液培养鲍曼复合群不动杆菌阳性、外周血巨细胞病毒阳性、G试验提示真菌感染阳性1例(例10);病原学检查为阴性者3例。
确诊后8例患儿定期IVIG替代治疗,随访至今感染频次显著减少,无后遗症。3例患儿未进行定期治疗,仍常有感染,其中1例伴有癫痫,另1例因反复腹泻继发巨幼红细胞贫血。
2.4 BTK突变分析
患儿基因检测结果见表2。剪接突变4例,错义突变3例,无义突变2例,缺失突变2例。基因突变涉及内含子2例,外显子9例。外显子分别为外显子18(3例),外显子15~17(2例)系同胞兄弟,外显子10(3例),外显子8(1例)。11例患儿中10例突变来源为母亲,1例为自发突变。例9患儿基因突变c.1908+1G>C为新发突变,该突变在HGMDpro数据库中未见报道。
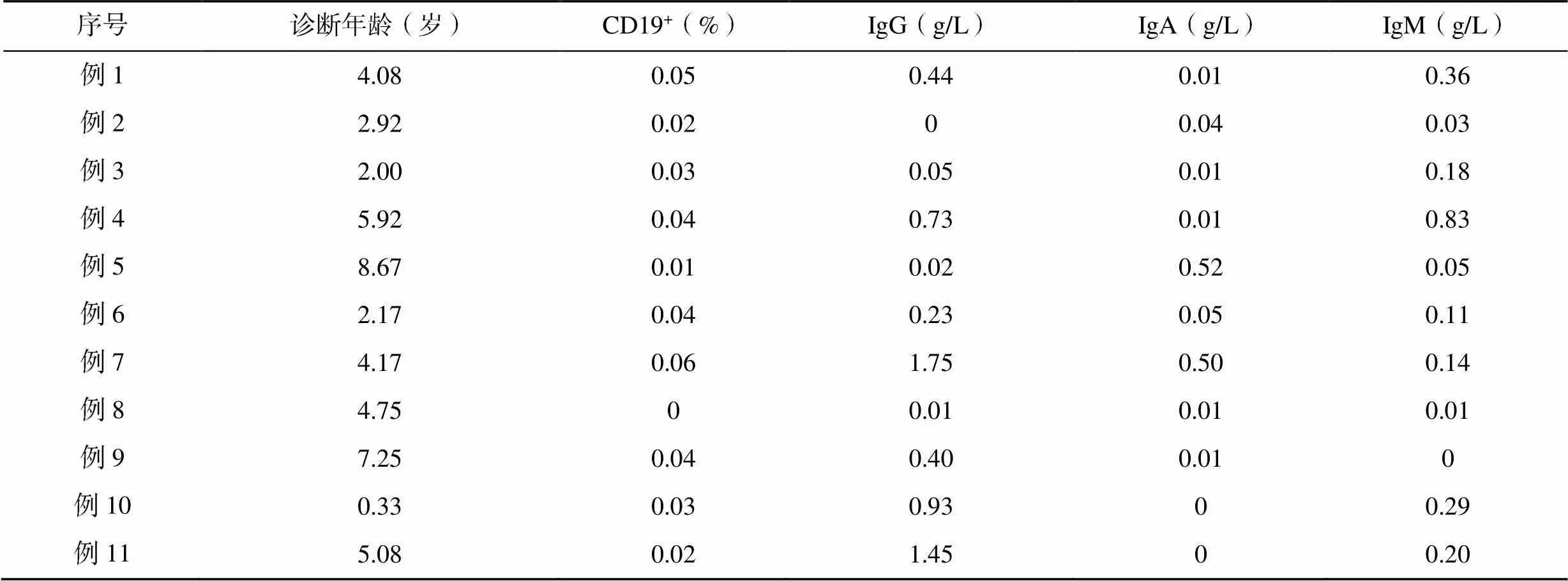
表1 11例患儿淋巴细胞亚群及体液免疫检测结果
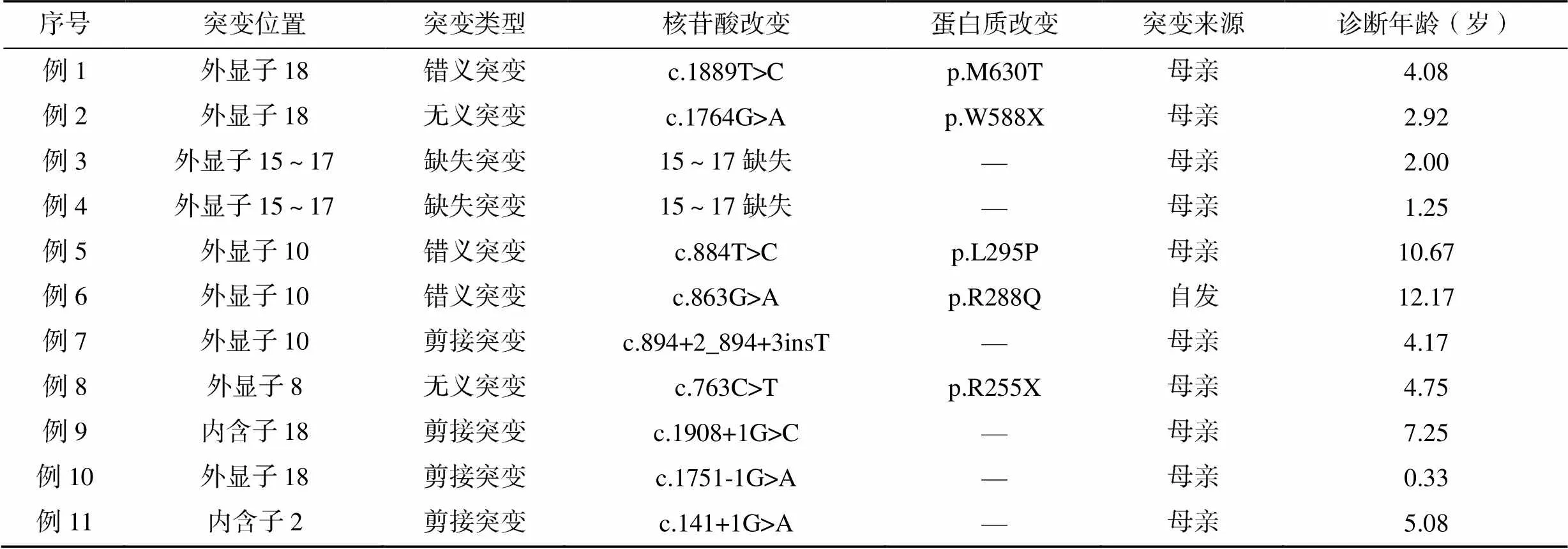
表2 XLA患儿BTK基因检测结果
2.5 SARS-CoV-2感染情况调查
对11例患儿进行SARS-CoV-2感染情况调查,共收到8例患儿的调查结果。4例患儿接种灭活疫苗,4例患儿未接种疫苗;截止到2023年1月,3例患儿感染SARS-CoV-2,均为轻型,按时输注丙种球蛋白,已康复,另5例患儿至今尚未感染SARS-CoV-2。
3 讨论
XLA是抗体缺陷病中最常见的一种,在原发性免疫缺陷病中的比例可达6%~11%。XLA的发病率因国家而异,国内尚未有XLA发病率的报道。与其他免疫缺陷病相比,XLA发病年龄相对较小,临床症状通常发生在出生后6个月至2岁[5];一旦母源性抗体从婴儿的血液中消失,将易发生呼吸道和消化道感染。国外研究报道XLA的平均确诊年龄为26个月。李瑛等[6]研究显示XLA患者基因确诊年龄为26~168月龄;陆红宇[7]对23例XLA患者研究发现其确诊年龄为24~164月龄;Chen等[8]对国内174例XLA患者的统计发现,其发病中位年龄1岁,平均诊断年龄为0.17~19.00岁。本研究中所有患儿均来自云南省,其中2例为彝族,患儿发病年龄0.25~6.00岁,中位年龄1.50岁,基因确诊年龄0.33~12.00岁,与国内报道相符。陈欢[9]研究发现具有较高IgG水平的患儿,近一半年龄超过12岁,本研究中患儿年龄均较小,年龄较大的2例患儿IgG水平亦较低。年龄最小的患儿在3个月时即出现多重病原体感染,其IgG<2g/L,IgM及IgA水平均很低。
XLA临床以反复化脓性感染最为常见,感染部位主要包括呼吸系统、消化系统及中枢神经系统。约90%的患儿会反复出现呼吸道感染,以流感嗜血杆菌、肺炎链球菌多见,其次是金黄色葡萄球菌和假单胞菌属细菌[10]。本研究中以呼吸道感染为主,其次是中枢神经系统感染及血流感染;呼吸道感染中最常见的病原体为肺炎链球菌,部分患儿甚至有多重病原体感染。年龄最小的患儿在3个月因严重肺炎入院,有多重病原体感染,包括巨细胞病毒、真菌及鲍曼不动杆菌。另有1例患儿(例2)脑脊液、骨髓及外周血均培养出木糖氧化无色杆菌反硝化亚种。巨细胞病毒及木糖氧化无色杆菌等病原体感染易发生于免疫功能低下者[11]。因此,对感染这些病原体的患者,临床医生应及时行免疫功能检测,及早筛查免疫缺陷病。除感染外,患儿还同时伴有长期腹泻、生长发育迟缓、贫血、关节炎、癫痫等疾病,其中生长发育迟缓是除感染外另一常见临床症状。
由于B淋巴细胞发育受阻,部分XLA患儿接种疫苗时对菌苗抗体接种反应极低或缺如,部分也会在血型鉴定时检测不到相应的血型抗体。本研究中1名患儿(例1)已接受两次风疹麻疹疫苗接种,但仍感染麻疹,且其血型用常规方法无法鉴定。XLA临床症状除常见的感染外,还有一些少见的并发症,如炎症性肠炎、肠道病毒性疾病、大颗粒淋巴性疾病、关节炎、慢性皮肤溃疡及疫苗相关性麻痹性脊髓灰质炎等,甚至可发展为胃肠道肿瘤[1]。
目前,世界范围内报道的基因突变类型已超过760多种,包括错义突变、插入、缺失、无义突变和剪接位点突变等,其中以错义突变最常见[6,8]。本研究中突变类型有剪接突变、错义突变、无义突变、缺失突变,其中以剪接突变为主,这可能与本组研究病例较少有关。目前BTK基因突变与临床严重度之间是否存在基因型-表型关系仍未明确,但有研究发现,错义突变的患儿起病年龄明显晚于其他突变类型的患儿[9,12]。本研究中确诊年龄最大的2例患儿均为10号外显子发生错义突变,初诊时其Ig水平均较低,与结论一致。最小患儿基因突变类型为18号外显子发生剪接突变,因重症肺炎就诊入院,初诊时其Ig水平均较低,经过定期注射丙种球蛋白,随访至今情况良好。本研究中2例患儿为彝族,其突变类型为错义突变和无义突变,都是常见突变类型,但其中1例无家族史,为自发突变。本研究发现1例新发突变(例9),突变位点为内含子18上发生剪接突变c.1908+1G>C,初诊时以中枢神经系统感染入院,并伴有癫痫。
XLA患儿因免疫缺陷是SARS-CoV-2感染的高危人群。本研究调查其中8例XLA患儿,仅4例患儿接种疫苗,均为灭活疫苗;截止到2023年1月,3例患儿感染SARS-CoV-2,均为轻型,按时输注丙种球蛋白感染已恢复。欧洲免疫缺陷病协会建议原发性抗体缺乏患者应接受疫苗接种,疫苗类型可为灭活疫苗或mRNA疫苗[13]。Salinas等[14]观察原发性免疫缺陷的患者接种疫苗后的免疫反应,XLA患者无抗体应答证据,但多数患者对疫苗接种有显著的T细胞反应,这可能为XLA患者提供保护。调查中3例已感染SARS-CoV-2的XLA患儿病情较轻,可能与接种疫苗及按时输注丙种球蛋白相关。目前没有直接的预后工具可用作XLA患者在感染SARS-CoV-2时的风险分层,表明需要对XLA患者的SARS-CoV-2感染进行有针对性的个体化临床评估,以改善结局并缩短住院时间。总的来说,XLA患者发生重症感染的风险增加,尤其是在Ig替代不足和(或)存在慢性肺部变化的情况下[15]。
目前,XLA的首选治疗方案为IVIG替代治疗,其可显著提高患儿血清IgG的水平,控制大多数患儿的感染症状,降低患者的死亡率及细菌感染率。普遍认为患儿血清IgG水平维持在6~8g/L可明显减少感染次数及减轻感染严重程度,IgG>8g/L可避免发生支气管扩张[16]。
综上,对婴幼儿时期即发生严重感染或反复感染免疫功能低下患儿,应及时进行淋巴细胞亚群及Ig的检测,并进行基因确诊。早期诊断,规范治疗,可减少感染及并发症的出现。
[1] EL-SAYED Z A, ABRAMOVA I, ALDAVE J C, et al. X-linked agammaglobulinemia (XLA): Phenotype, diagnosis, and therapeutic challenges around the world[J]. World Allergy Organ J, 2019, 12(3): 100018.
[2] AHMED A, LIPPNER E, KHANOLKAR A. Clinical aspects of B cell immunodeficiencies: The past, the present and the future[J]. Cells, 2022, 11(21): 3353.
[3] CONLEY M E, NOTARANGELO L D, ETZIONI A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies)[J]. Clin Immunol, 1999, 93(3): 190–197.
[4] 陈同辛, 王玺. 原发性免疫缺陷病诊断标准[J]. 实用儿科临床杂志, 2006, 21(9): 573–576.
[5] LEDERMAN H M, WINKELSTEIN J A. X-linked agammaglobulinemia: An analysis of 96 patients[J]. Medicine (Baltimore), 1985, 64(3): 145–156.
[6] 李瑛, 张翠, 杨颖, 等. X-连锁无丙种球蛋白血症临床分析及基因诊断[J]. 临床儿科杂志, 2019, 37(3): 192–195.
[7] 陆红宇. 23例X连锁无丙种球蛋白临床分析[D]. 重庆: 重庆医科大学, 2015.
[8] CHEN X F, WANG W F, ZHANG Y D, et al. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: Report from Shanghai, China (2000-2015)[J]. Medicine (Baltimore), 2016, 95(32): e4544.
[9] 陈欢. 93例X连锁无丙种球蛋白血症的临床表现及基因分析[D]. 重庆: 重庆医科大学, 2019.
[10] 殷勇, 袁姝华. 儿童X-连锁无丙种球蛋白血症[J]. 中华实用儿科临床杂志, 2018, 33(4): 288–291.
[11] 中国医师协会新生儿科医师分会, 中国医师协会新生儿科医师分会感染专业委员会, 中华新生儿科杂志编辑委员会. 新生儿巨细胞病毒感染管理专家共识[J]. 中华新生儿科杂志, 2021, 36(6): 1–7.
[12] 王莹, 应文静, 孙金峤, 等. 中国X连锁无丙种球蛋白血症40例基因型表型相关性分析[J]. 中国循证儿科杂志, 2012, 7(1): 4–10.
[13] FRIEDMANN D, GOLDACKER S, PETER H H, et al. Preserved cellular immunity upon influenza vaccination in most patients with common variable immunodeficiency[J]. J Allergy Clin Immunol Pract, 2020, 8(7): 2332–2340.
[14] SALINAS A F, MORTARI E P, TERRERI S, et al. SARS-CoV-2 vaccine induced atypical immune responses in antibody defects: Everybody does their best[J]. J Clin Immunol, 2021, 41(8): 1709–1722.
[15] RISE N, TOUBORG T, LUNDSTED D H, et al. Case report: Evolution of pulmonary manifestations and virological markers in critical COVID-19 infection in Bruton’s agammaglobulinemia[J]. Front Immunol, 2022, 13: 1057065.
[16] 中华医学会儿科学分会免疫学组, 《中华儿科杂志》编辑委员会. 原发性免疫缺陷病抗感染治疗与预防专家共识[J]. 中华儿科杂志, 2017, 55(4): 248–255.
Clinical analysis of 11 cases of X-linked agammaglobulinemia
XIA Shimei, YUAN Mei, FENG Xingxing, JIANG Hongchao, DUAN Shaoqin, LI Weixi, KUI Liyue, ZHOU Bailing
1.Department of Clinical Laboratory, Kunming Children’s Hospital, Kunming 650100, Yunnan, China; 2.Department of Infectious Disease, Kunming Children’s Hospital, Kunming 650100, Yunnan, China; 3.Department of Hematology, Kunming Children’s Hospital, Kunming 650100, Yunnan, China; 4.College of Chinese Medicine, Yunnan University of Chinese Medicine, Kunming 650100, Yunnan, China
To analyze the clinical features of children with X-linked agammaglobulinemia (XLA) and the gene mutation of Bruton’s tyrosine kinase (BTK) in Yunnan region.The clinical data of 11 children with XLA diagnosed by genetic testing in Kunming Children’s Hospital from January 2018 to June 2022 were retrospectively analyzed, and the mutation of BTK gene was analyzed by Sanger sequencing.All of the 11 children were male. The attack age range was from 0.25 to 6.00 years old, and the median age was 1.50 years old. The age at diagnosis range was from 0.33 to 12.00 years old, and the median age was 4.17 years old. The main clinical manifestations were respiratory tract infection and most children had severe infections, followed by growth retardation. The immunoglobulin level was low in all children, and the percentage of peripheral blood CD19+B lymphocytes was almost absent. The results of genetic testing showed 4 splicing mutations, 3 missense mutations, 2 nonsense mutations, and 2 deletion mutations. One new mutation was found and the mutation site was c.1908+1G>C.For boys with severe infections and slow growth in infancy, genetic testing should be performed as soon as possible.
X-linked agammaglobulinemia; Bruton’s tyrosine kinase; Primary immunodeficiency
R715
A
10.3969/j.issn.1673-9701.2023.25.001
国家自然科学基金项目(81960780);昆明市卫生科技人才培养项目(2020-SW-118)
周百灵,电子信箱:79769522@163.com
(2023–03–20)
(2023–08–22)



