傅亚瑜,金 珊,刘康文,方 向
(1.安徽中医药大学,安徽 合肥 230038;2.安徽中医药大学第一附属医院,安徽 合肥 230031)
Wilson病是一种因ATP7B基因突变而引发的单基因隐性遗传病。基因突变导致铜排泄障碍,过量的铜主要沉积在肝、脑、肾、角膜等组织器官,因此出现相应的症状、体征。Wilson病根据临床表型的不同可以表现为神经系统、消化系统、内分泌系统、皮肤系统、心血管系统及泌尿系统等多系统的损害[1]。研究[2]发现Wilson病患者中有29.2%~52.0%人群合并肾损害,临床上常见的肾损害主要表现为:血尿、蛋白尿、水肿,严重时可出现急慢性肾炎、肾病综合征、IgA肾病等,其中IgA肾病极为少见。目前国外已经报道过的wilson病相关IgA肾病仅有3例,其中2例见于儿童,另1例是一名20岁的日本患者[3],国内统计则不完全。因此,在这里笔者将报道1例Wilson病相关的IgA肾病。
1 病历摘要
本次研究经过本院医学伦理委员会同意。患者男18岁,于2011年5月不明原因下出现眼睛、皮肤发黄,全身乏力,渐出现腹部胀大、双下肢肿胀,食欲不佳,就诊于当地市级医院,查铜蓝蛋白低,尿铜高,KF环(+),诊断为“肝豆状核变性”,后于2012年~2019年间多次入住于我院,定期予排铜、保肝、护脑等治疗,期间病情控制尚可。2019年8月,患者因“双下肢水肿2周”再次收住我院。查体:神志清楚,精神可,语言尚清晰,伸舌居中,双侧瞳孔等大等圆,直径2.5 mm,KF环(+),四肢肌力、肌张力尚可,生理反射(++),病理征(-)。腹部稍膨隆,双下肢重度凹陷性水肿。实验室检查发现:天门冬氨酸氨基转移酶 48U/L,白蛋白16.9 g/L,碱性磷酸酶219 U/L;尿蛋白 (+++),尿隐血(+++),管型总数:4.22 μl,病理管型:1.84 μl,尿红细胞 4 419;24 h尿蛋白定量:6.16 g/24 h;尿总蛋白:7 705 mg/L;尿免疫球蛋白G 156 mg/L,尿转铁蛋白62.40 mg/L,尿微量白蛋白1 360 mg/L,尿α1微球蛋白22.10 mg/L,尿β2微球蛋白 0.27 mg/L; 24 h尿铜 455.68 μg;免疫球蛋白M未见异常,免疫球蛋白A 4.76 g/L,免疫球蛋白G 6.11 g/L,补体C3 0.64 g/L;肝胆胰脾彩超:1.肝豆状核变性肝病;2胆囊壁水肿;3.大量腹水;肾脏输尿管膀胱彩超:双肾皮质回声稍增粗增强,皮髓质境界稍模糊,提示肝豆状核变性肾病;肾穿后各项检查如下:①免疫荧光学检查:可见4个肾小球,各免疫复合物沉积强度分别为:IgA(+++),IgM(+++),IgG(-),C3(++)。②光镜检查:镜下可见1个细胞纤维性新月体形成,其余肾小球系膜细胞和基质轻度弥漫增生,局灶节段加重,系膜区可见嗜复红蛋白沉积。③电镜检查:肾小球系膜细胞和基质增生,系膜区大块状、高密度电子致密物沉积,上皮细胞足突节段性融合。④肾穿病理结果:结合临床,符合轻度系膜增生型IgA肾病。见图1。
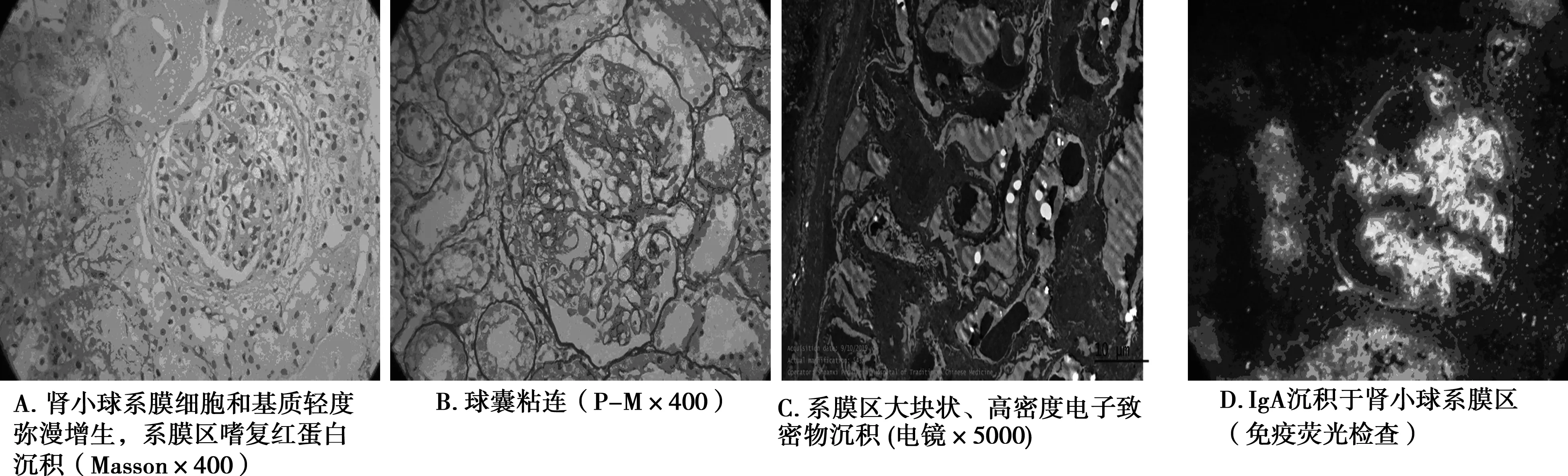
图1 各项检查结果
2 讨论
临床上Wilson病患者相关的肾损害大致可分为原发性及继发性两种。其中原发性肾损害主要见于因该病自身原因导致的铜沉积于肾脏或该病导致IgA及其免疫复合物沉积于肾小球系膜区,进而出现的肾损害;继发性肾损害则主要见于Wilson病治疗过程中青霉胺导致的药物性肾损害。其主要发病机制如下:(1)原发性肾损害:①铜沉积:人体主要依靠ATP7B维持铜的稳态平衡。ATP7B基因突变,导致编码的ATP7B异常,使得经肠道吸收入血后的铜无法转运至高尔基体内与血浆蛋白结合形成铜蓝蛋白,大量的铜沉积于体内[4]。Bickel等人曾报道Wilson病患者肾组织内的含铜量超出正常人的10~20倍。Wolff、Reynolds等人先后发现肾小管上皮细胞、肾小球囊壁层上皮细胞均可见铜粒沉积。此外,铜离子还能通过激活脂质过氧化反应致使自由基受损、影响细胞能量代谢,进一步使得肾小管重吸收功能下降,进而出现肾小管酸中毒、高钙尿症及肾结石、肾性糖尿、氨基酸尿等临床表现[5]。②IgA及其免疫复合物沉积:Wilson病患者可因基因突变导致铜代谢障碍,随着疾病进展铜颗粒不断沉积于肝脏,导致肝功能异常、肝脏的合成能力减弱,合成的血清蛋白量减少,肝内的浆细胞及骨髓代偿性增生,γ-球蛋白增多,与此同时肝脏的生物转化功能下降,对IgA及IgA复合物的清除力下降,IgA及其免疫复合物随着血液循环不断蓄积于肾小球系膜区,日久导致IgA肾病的出现[6]。临床上IgA肾病主要表现为无症状的镜下血尿或发作性血尿,可能伴有蛋白尿、肾功能不全或高血压等,需尽早进行肾穿刺以明确诊断及病理分级。(2)继发性肾损害:青霉胺是目前我国临床上用于排铜的一线药,可有效缓解Wilson病患者的临床症状。但该药也可能导致诸多不良反应,据统计[7],约有30%的Wilson病患者因长期服用青霉胺导致各种严重不良反应而停药,其中肾损害是其主要的药物不良反应之一。长期使用青霉胺可能导致肾小管受损,临床上常表现为蛋白尿、肾病综合征,也有间质性肾炎、膜性肾病、肾衰竭、ANCA相关的新月体肾小球肾炎的报道[8-10]。
临床上对于Wilson病相关的肾损害首先需分清原发性或继发性,再明确是由于铜沉积还是IgA及其复合物沉积所致,最后针对病因采取安全、高效的应对措施,阻断Wilson病相关肾损害的进展。关于Wilson病相关的肾损害,一般可从以下几个方面进行鉴别:(1)肾脏病变相关指标:在对70例已确诊为Wilson病的患者及70例无血缘关系的健康人群进行肾脏病变相关指标检测发现,在原发性铜沉积型肾损害中,因铜颗粒既能沉积于肾小管,又能沉积于肾小球,因此既有反映肾小管损害的β2-MG(β2-微球蛋白)、LMW(低分子量蛋白)的指标升高也有反映肾小球损害的尿ALG(白蛋白)、HMW(高分子量蛋白)的指标升高;而在原发性IgA及其免疫复合物沉积型肾损害中,因IgA及免疫复合物一般仅沉积于肾小球系膜区,因此仅有反应肾小球损害的尿ALG、HMW的指标升高[11]。(2)用药情况:根据患者既往或现在有无青霉胺的使用史,较为容易鉴别。青霉胺继发的肾损害一般与用药时间、剂量密切相关,临床上多表现为蛋白尿、超敏C-反应蛋白升高。这种继发性肾损害多在用药后的2个月~2年出现,蛋白尿出现的概率与用药时间呈正相关,当用药时间由6个月延长至1年时,其继发肾损害的概率也由2%增长至30%。此外,青霉胺用量超出250~500 mg/d时,更易出现蛋白尿[12]。因青霉胺继发的肾损害必要时需调整剂量乃至停药或加用激素。(3)病理表现:①铜沉积:肾近端、远端小管上皮细胞、肾小球囊壁层上皮细胞均可见铜粒沉积。早期可见近端小管上皮细胞扁平,刷状缘消失,肾近端小管基底膜增厚,间质内可见纤维化体局限性白细胞浸润,晚期则以肾小管硬化及毛细血管闭塞的病理表现为主[13-14]。②IgA及其免疫复合物沉积:免疫荧光学检查发现肾小球系膜区可见IgA、IgM、补体C3等的异常沉积,且以IgA为主,同时肾小球可见颗粒样或团块样沉积;光镜、电镜可见肾小球系膜细胞和基质增生,系膜区大块状、高密度电子致密物沉积;③继发性肾损害:因使用青霉胺而继发的肾损害病理上主要表现为膜性肾小球肾炎,少部分表现为ANCA相关性肾炎、肾衰竭等[15]。
本研究病例中,患者既往以肝脏损害为主要表现,动态随访无肾功能损害证据,无青霉胺使用史,无感染、发热、腹泻等常见肾功能损害诱因,本次发现患者大量蛋白尿,入院后完善免疫荧光学、光镜及电镜检查发现符合轻度系膜增生型IgA肾病,请相关科室会诊后,予激素治疗,症状明显改善。因此临床上Wilson病患者如果出现肾损害的相关临床表现及实验室检查证据后,应尽快行进一步检查,必要时行肾脏穿刺,明确发病原因究竟为原发性还是继发性,若为原发性,明确是铜沉积型还是IgA及其免疫复合物沉积型,避免误诊、延误最佳治疗时机。



