张苏闽 汤晓枢 周媛媛(上海长城药业有限公司 上海 201707)
脑蛋白水解物原料肽含量测定方法的改进
张苏闽汤晓枢*周媛媛
(上海长城药业有限公司上海201707)
目的:优化脑蛋白水解物原料肽含量测定方法。方法:采用Agilent Zorbax Eclipse-AAA色谱柱(4.6 mm×150 mm, 5 mm), 流动相A:0.04 mol/L磷酸盐缓冲液(pH 7.80), 流动相B:乙腈-甲醇-水(45∶45∶10), 流速1.5 ml/min, 柱温40 ℃, 检测波长:338 nm, 262 nm, 梯度洗脱。结果:各种氨基酸的浓度在15.0~120.0 mg/ml范围内与峰面积呈良好的线性关系(r>0.999), 各种氨基酸的平均回收率为98.1%~103.6%。结论:该方法快速、简便、专属性强、重复性好, 结果准确可靠, 适用于脑蛋白水解物原料肽含量的测定。
脑蛋白水解物原料肽含量测定氨基酸高效液相色谱
脑蛋白水解物原料(国药准H31023028)为我们公司原料药车间生产的原料药,系健康猪的鲜大脑经脱脂和水解而成。2013年6月28日国家药典委员会发布了《关于脑蛋白水解物原料国家标准的公示稿》,明确了需进行脑蛋白水解物原料肽含量的测定,即需对脑蛋白水解物原料中游离及水解氨基酸含量进行测定,并用于计算肽含量(肽含量=水解氨基酸含量-游离氨基酸含量)。
大多数氨基酸不含有芳香环等生色团,无紫外吸收,需要先将氨基酸衍生为具有较强紫外或荧光吸收的衍生物[1]。氨基酸HPLC柱前衍生化的分析方法有多种,以Waters AccQ·Tag衍生法[2-4]、OPA-FMOC(邻苯二甲醛—9-芴甲基氯甲酸酯)衍生法测定氨基酸最为常用[5-7]。本公司曾用OPA-FMOC衍生法测定脑蛋白水解物原料中游离及水解氨基酸含量(色谱柱Hypersil ODS-C18:4.6 mm×100 mm,5 mm,流动相A:pH 7.20的醋酸盐缓冲液,流动相B:pH 7.20的醋酸盐缓冲液-乙腈-甲醇,梯度洗脱)。但我们发现该种色谱条件在分析检测氨基酸时存在以下问题:①在同时分离16种氨基酸时,部分氨基酸分离度差,系统适用性不佳;。②测定方法的重现性差;③进行梯度洗脱的A、B相盐浓度高,直接影响色谱柱的使用寿命;④该系统对pH较敏感,配制A、B相pH误差会直接影响氨基酸的分离。本研究参考文献优化了脑蛋白水解物原料肽含量测定的方法[8]。
1 仪器与试药
Agilent 1100液相色谱仪,DAD检测器、自动进样器(安捷伦科技中国有限公司)。Sartorius BP121S电子天平(赛多利斯科学仪器北京有限公司)。16种氨基酸对照品(批号:140624-200805,中国药品生物制品检定所)。脑蛋白水解物原料(批号:20101102)为本公司产品。甲醇、乙腈为色谱纯,水为超纯水,磷酸氢二钠及其他试剂均为分析纯。
2 方法与结果
2.1色谱条件
Agilent Zorbax Eclipse-AAA色谱柱(4.6 mm×150 mm,5 mm),流动相为A:0.04 mol/L磷酸盐缓冲液(pH 7.8)(取磷酸二氢钠(NaH2PO4·2H2O)6.24 g,加水1 000 ml溶解,用5 mol/L的氢氧化钠溶液将pH调至7.80±0.05);流动相B:乙腈-甲醇-水(45∶45∶10),按表1进行梯度洗脱。柱温:40 ℃。检测波长:338 nm,262 nm。流速:1.5 ml/min.。

表1 梯度洗脱
2.216种氨基酸对照品溶液的配制
对照品溶液的制备:称取16种氨基酸对照品各约30 mg,至100 ml容量瓶中,以0.04 mol/L盐酸溶液溶解至刻度(对照品贮备液),再精密量取2 ml,至10 ml容量瓶中用水稀释至刻度,摇匀,备用。
2.3供试品溶液的配制
游离氨基酸含量测定供试品溶液:精密称重脑蛋白水解物原料300 mg,置100 ml容量瓶中,加水溶解并超声10 min,加水稀释至刻度,滤过,取续滤液作为游离氨基酸含量测定供试品溶液。
水解氨基酸含量测定供试品溶液:精密称取脑蛋白水解物原料200 mg,置硬质安瓿中,加盐酸2 ml,摇匀,充氮封口,于110 ℃水解20 h,放冷,启封后溶液置蒸发皿中,水浴蒸发至干,加水使残留物溶解并稀释至100 ml,作为水解氨基酸含量测定供试品溶液。
2.4衍生化程序
采用Agilent液相色谱仪自带的柱前衍生的方法,设定进样程序,对所得的氨基酸样品进行衍生化反应,进样器程序设定见表2。
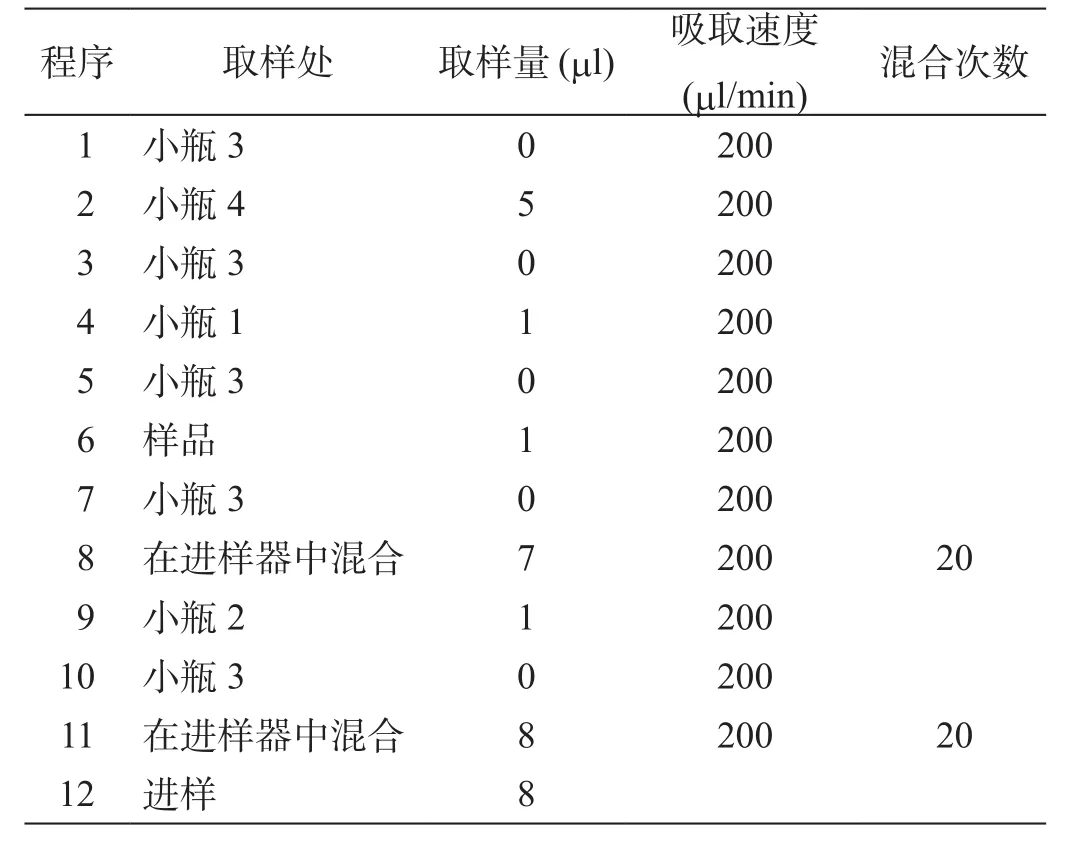
表2 衍生进样程序设定
氨基酸衍生试剂配制方法:
小瓶 1:8 mg/ml邻苯二甲醛(OPA)溶液:精密称取80 mg邻苯二甲醛(OPA),依次加入7 ml硼酸缓冲液溶液、1 ml乙腈和125 ml 巯基丙酸,摇匀并溶解,即得。
小瓶 2:5 mg/ml 9-芴甲基氯甲酸酯(FMOC)溶液:精密称取50 mg 9-芴甲基氯甲酸酯(FMOC),用乙腈溶解并稀释至10 ml,摇匀并溶解,即得。
小瓶 3:双重蒸馏水。
小瓶 4:硼酸缓冲液:精密称取1.236 g硼酸至50 ml容量瓶中,加水超声至全部溶解,用40%氢氧化钠溶液调节pH至10.2,即得。
2.5方法学考察
2.5.1系统适用性试验
取需测定的16种氨基酸的对照品:门冬氨酸(Asp)、谷氨酸(Glu)、丝氨酸(Ser)、组氨酸(His)、甘氨酸(Gly)、苏氨酸(Thr)、精氨酸(Arg)、丙氨酸(Ala)、缬氨酸(Val)、甲硫氨酸(Met)、色氨酸(Trp)、异亮氨酸(Ile)、苯丙氨酸(Phe)、亮氨酸(Leu)、盐酸赖氨酸(Lys)、脯氨酸(Pro),依法配制对照品溶液,衍生后进样,进行系统适用性试验(图1)。
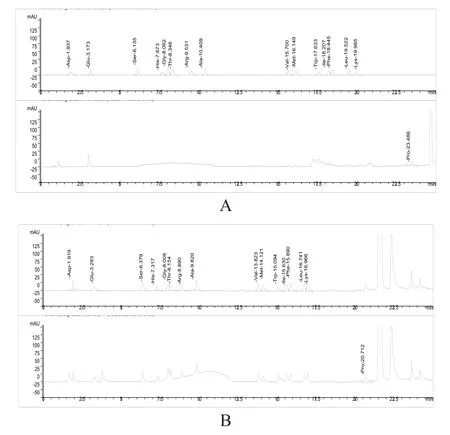
图1 色谱条件改进前后16种氨基酸系统适用性试验色谱图
从图1A可见,在该色谱条件下,需测定的16种氨基酸均能完全分离,峰形佳,较图1B中原色谱分离条件有了明显的改善。分离度数据显示(表3),改进前16种氨基酸的分离度未完全达到药典规定的分离度要求,缬氨酸与甲硫氨酸、亮氨酸与赖氨酸的分离度仅大于1.0,且甘氨酸与苏氨酸未完全分离。经色谱条件改进优化后各氨基酸的分离度均大于1.5,分离效果明显改善,实现了16种氨基酸在同一色谱条件下的完全基线分离。
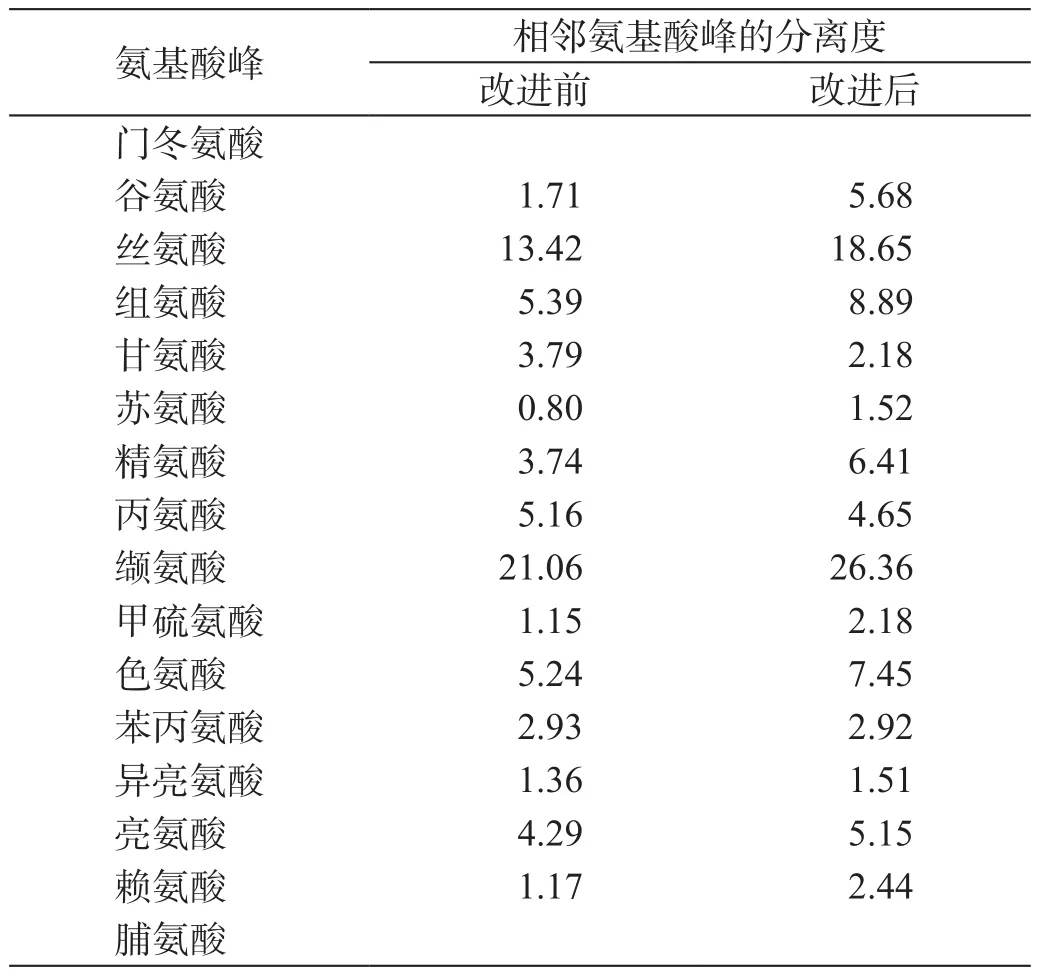
表3 16种氨基酸峰分离度
2.5.2线性范围
分别取16种氨基酸对照品配制成一系列浓度的混合对照品溶液,分别取上述对照品溶液衍生后进样,并计算16种氨基酸的线性回归方程(表4)。
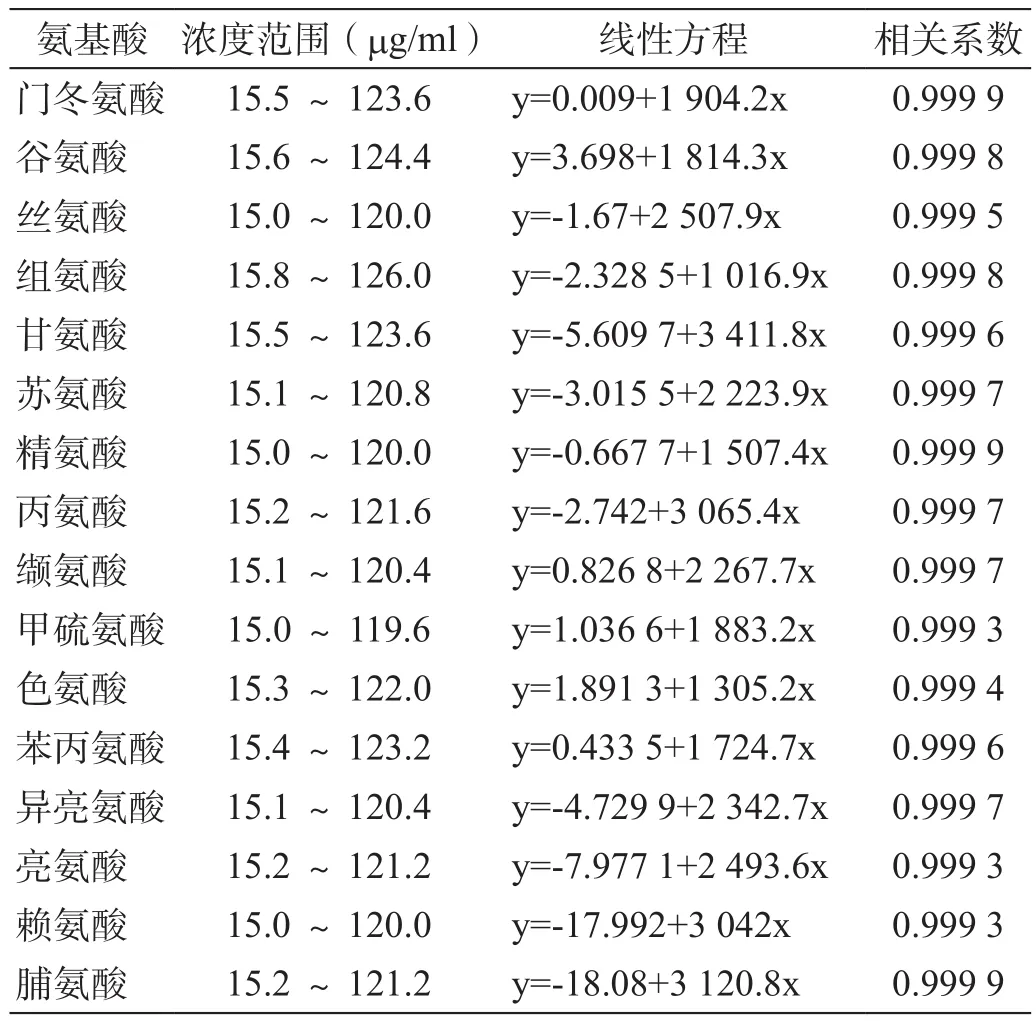
表4 16种氨基酸线性试验
结果显示,16种氨基酸的浓度在约15.0~120.0 mg/ml范围内与峰面积呈良好的线性关系(r>0.999)。
2.5.3精密度试验
分别取16种氨基酸对照品溶液衍生后连续进样6次,结果见表5。
连续进样6次16种氨基酸峰面积偏差的RSD均<2.0%。可知该色谱条件下氨基酸测定方法的精密度较好。
2.5.4重复性试验
取同一批供试品,依法制备游离氨基酸和水解氨基酸供试品溶液各6份,衍生后分别测定游离、水解氨基酸的含量,并分别计算脑蛋白水解物原料肽含量(表6)。
由表6结果可知:肽含量均值为419.5 mg/g,RSD小于2.0%,表明采用该方法测定脑蛋白水解物原料肽含量重复性较好,测定结果可靠。
2.5.5溶液稳定性试验
取制备的游离氨基酸和水解氨基酸供试品溶液,分别于0、2、4、8、12、18、24 h衍生并进样检测,以0 h进样样品的含量为100%,比较供试品溶液中各氨基酸的日内稳定性(表7)。
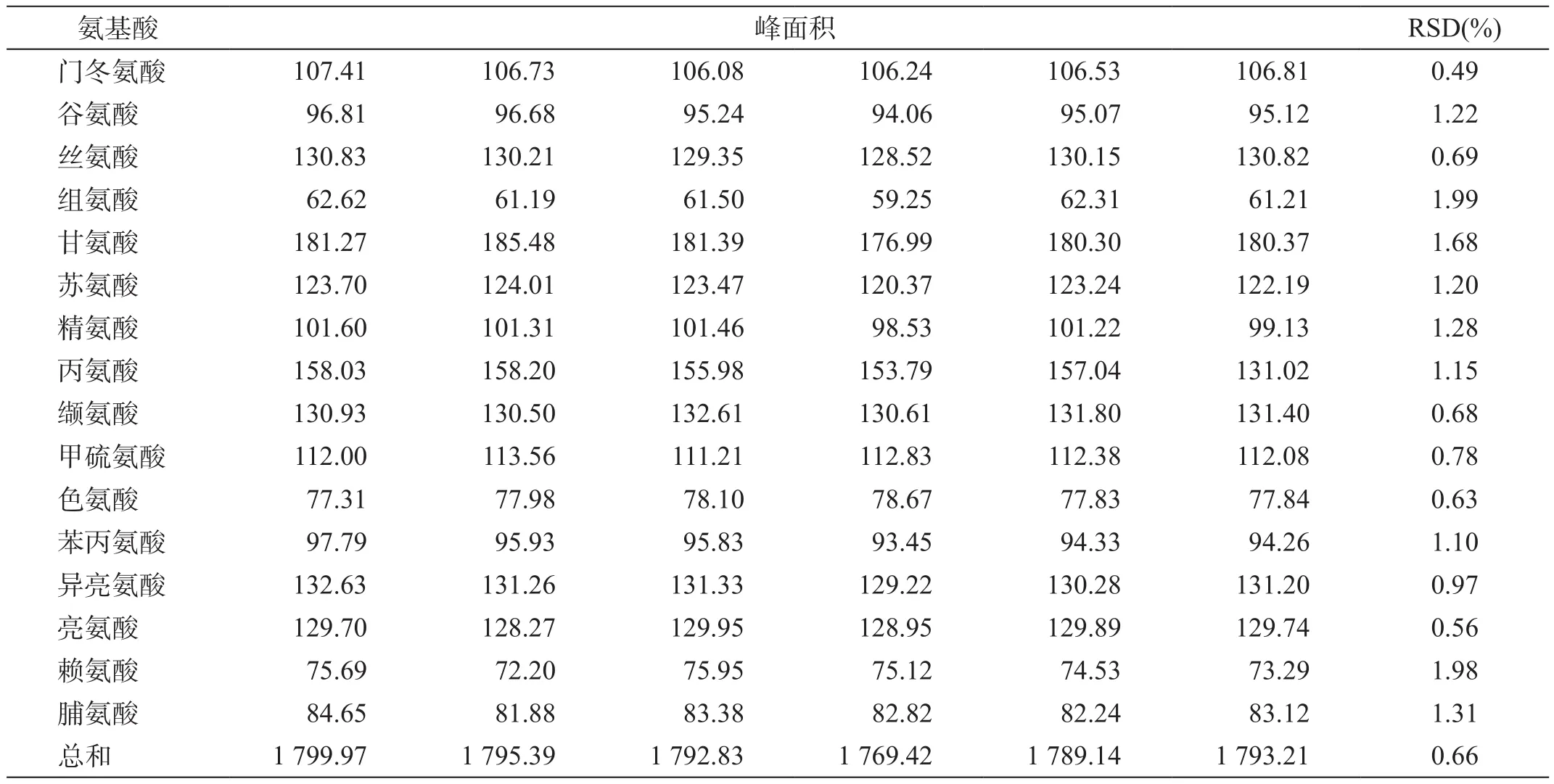
表5 脑蛋白水解物原料肽含量测定精密度试验
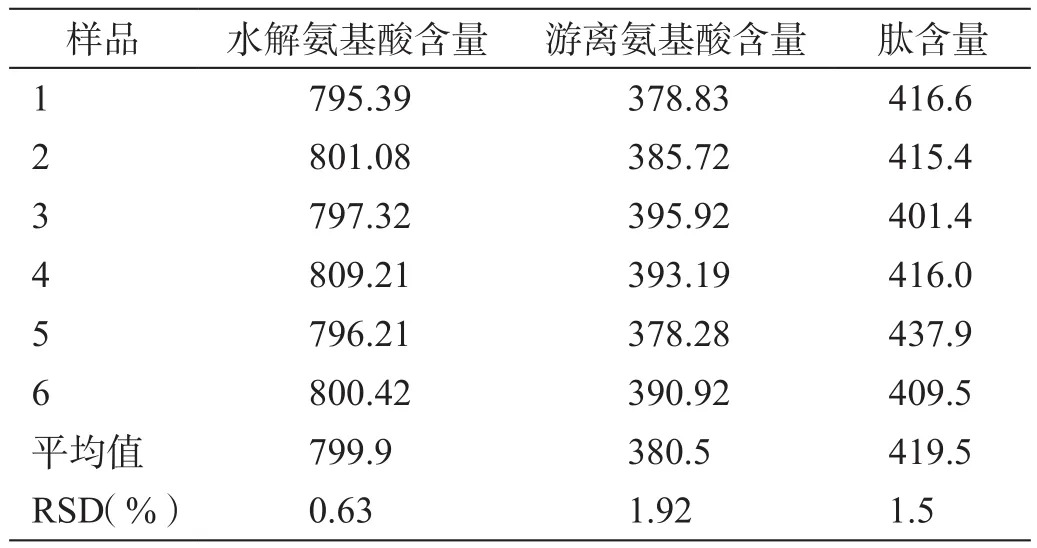
表6 脑蛋白水解物原料肽含量测定重复性试验(mg/g)
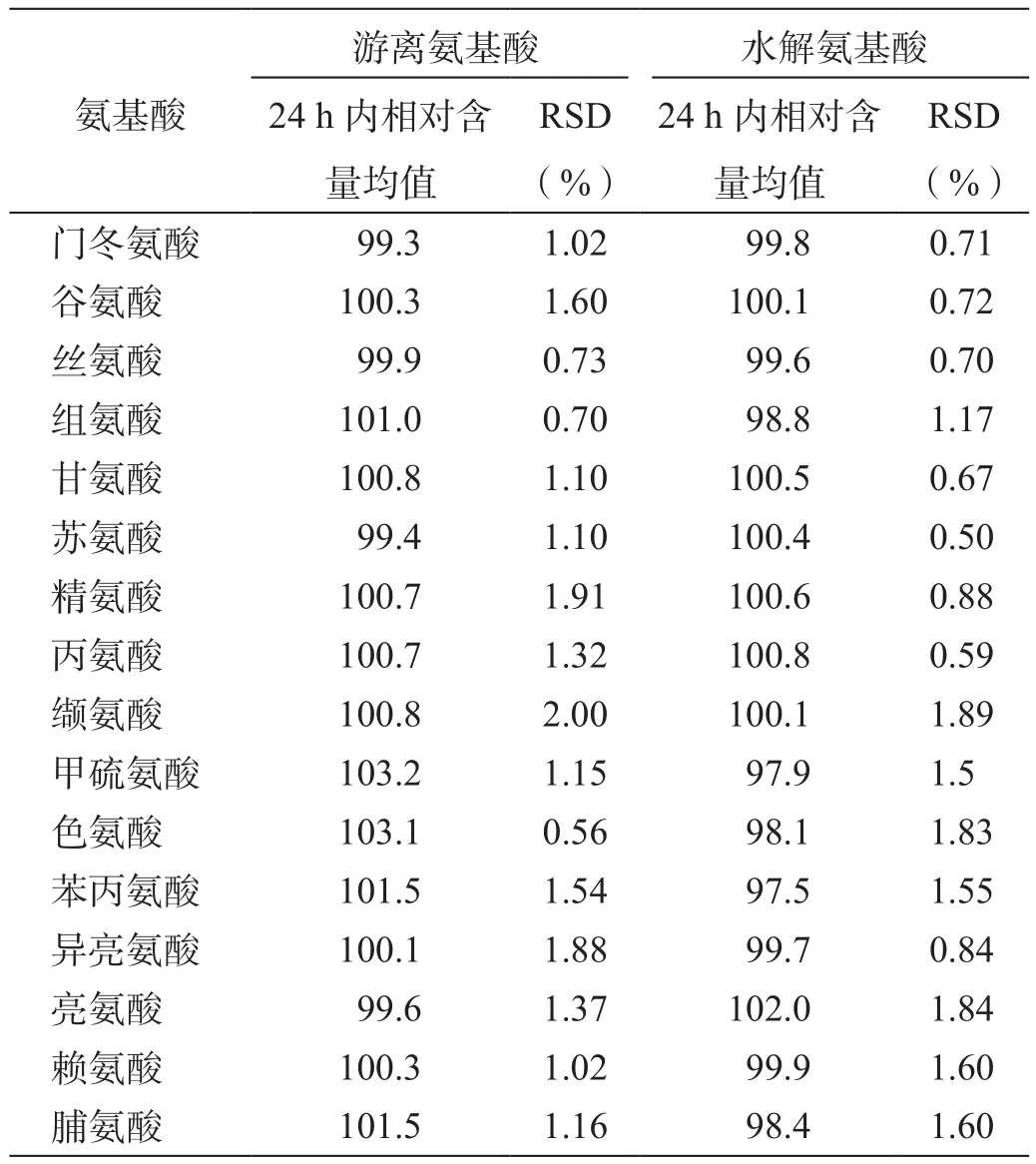
表7 脑蛋白水解物原料供试品溶液稳定性试验(%)
结果显示,游离和水解供试品溶液中所含16种氨基酸在24 h内的含量变化的RSD均小于2.0%,表明供试品溶液在24 h内稳定,含量测定结果可靠。
2.5.6加样回收试验
取供试品适量,按照高(120%)、中(100%)、低(80%)三个浓度加入含16种氨基酸的对照品储备液,依法制备加样回收的供试品溶液,每个浓度各3份。取上述加样溶液衍生后进样,并计算加样回收率(表8)。
结果表明,游离、水解氨基酸含量测定中16种氨基酸的回收率均在98.1%~103.6%范围内,RSD均小于2.0%,证明本方法回收率结果良好。
2.6样品测定
取不同批次的脑蛋白水解物原料,依法制备游离、水解供试品溶液,并衍生化进样。测定并计算肽含量(表9)。
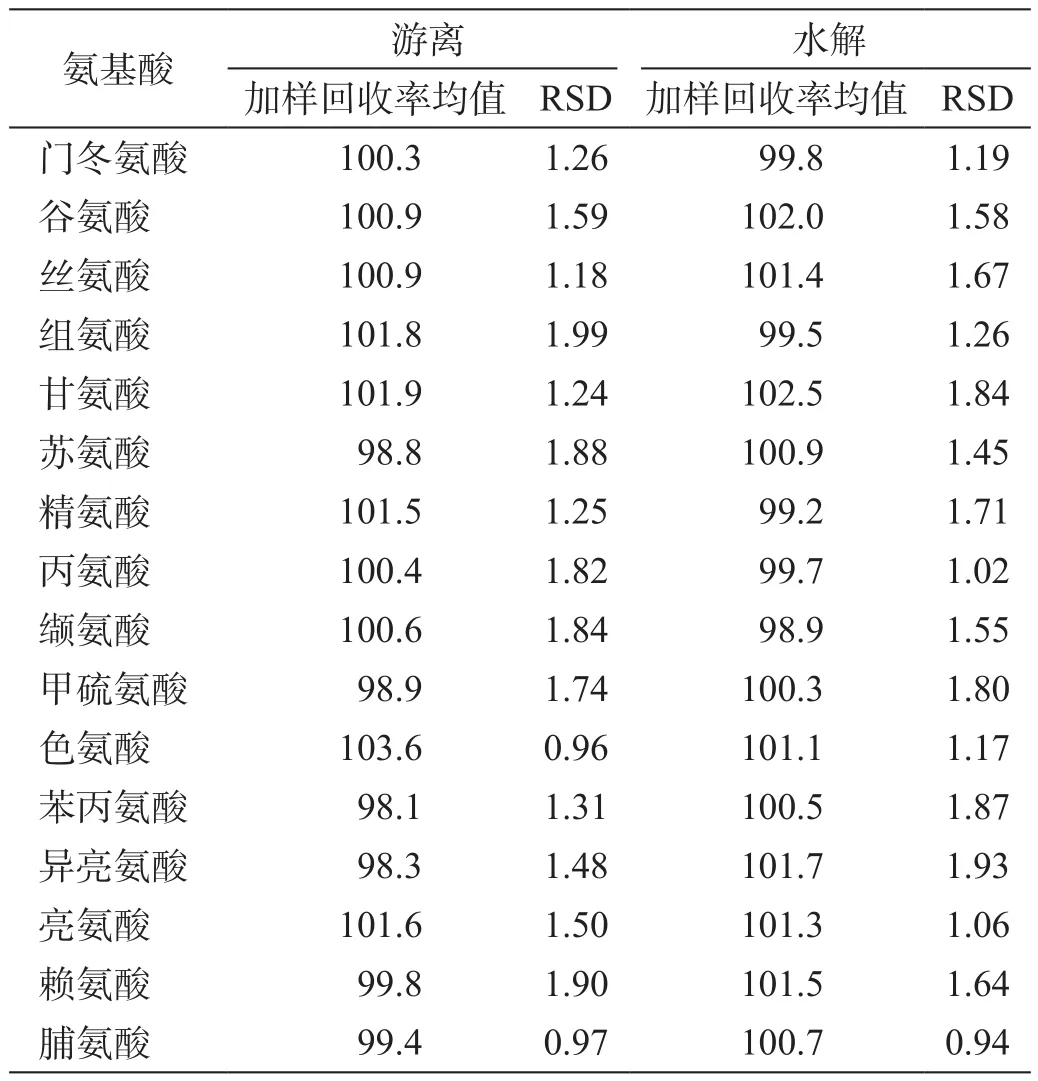
表8 脑蛋白水解物原料肽含量测定加样回收试验(%)
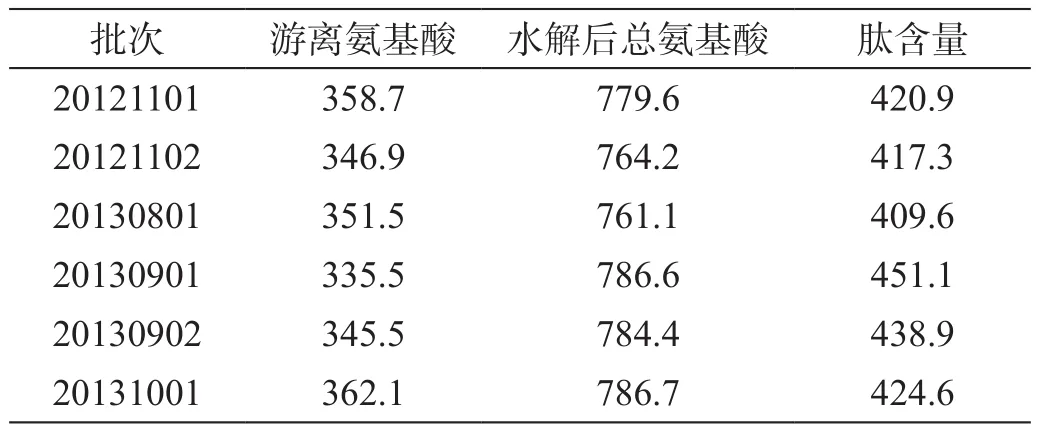
表9 脑蛋白水解物肽含量测定(mg/g)
3 讨论
3.1检测器的选择
OPA-FMOC衍生法测定氨基酸,其中一级氨基酸(Asp、Glu、Ser、His、Gly、Thr、Arg、Ala、Val、Met、Trp、Ile、Phe、Leu、Lys共15种)首先与OPA反应,形成OPA-氨基酸,在338 nm下有最大吸收。二级氨基酸(Pro)则与FMOC衍生生成FMOC-氨基酸,在262 nm下有最大吸收。因加入吲哚与3-巯基丙酸相结合降低了氨基酸的疏水性,所以OPA衍生化产物较FMOC衍生化产物色谱出峰时间较早。
在原有方法中,我们选用VWD检测器进行检测,并在一级氨基酸中最后出峰的盐酸赖氨酸(Lys)出峰后设置一个波长切换,检测波长由338 nm切换至262 nm,用于检测二级氨基酸的脯氨酸(Pro)。该方法对使用的紫外检测器要求不高,但因波长切换导致检测器的急速光栅变化,脯氨酸(Pro)出峰时检测器未必达到稳定的状态,且在波长切换时如基线发生抖动,会在图谱上形成一个较大的上升、下降平台,使得262 nm下脯氨酸检测数据不稳定。故我们建议采用DAD检测器进行两级氨基酸的检测,即分别连续收集338 nm和262 nm两个波长的信号,这样保证了一级、二级氨基酸同时检测的准确性。
3.2参比波长的设定
在Agilent Zorbax Eclipse-AAA色谱柱说明书中,明确规定了氨基酸检测参比波长的设定,参比波长的设定参数如下:338 nm:10 nm带宽(bw),参比波长:390 nm,20 nm带宽(bw)(适用于OPA-氨基酸);262 nm:16 nm带宽(bw),参比波长:324 nm,8 nm带宽(bw)(适用于FMOC-氨基酸)。峰宽:>0.03 min(0.5 s)。狭缝间距:4 nm。
在方法优化过程中我们进行了相关比对试验,对比了其与工作站默认参比波长设置的区别,结果显示采用这种参比波长的设定,对于氨基酸检测的图谱起到了明显的优化作用,设定后梯度洗脱条件下色谱图的基线较未设定前更平稳,不再有明显漂移。各氨基酸峰的对称性也有改善,峰型更佳。从而提高了多种氨基酸含量测定的准确性(图2)。
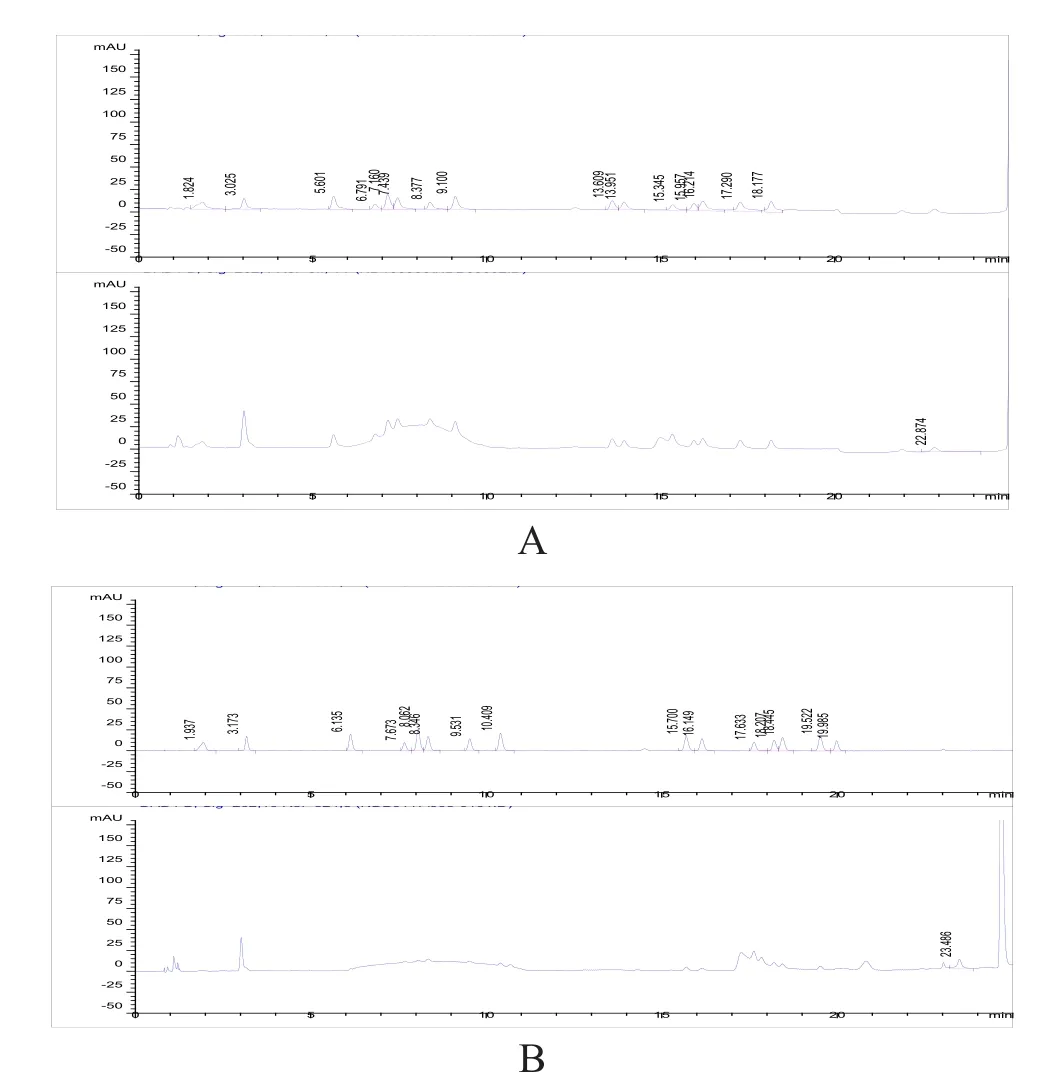
图2 参比波长优化前后16种氨基酸的分析图谱
3.3流动相的改进
原方法中使用的流动相存在以下问题:①流动相A为pH 7.20的醋酸盐缓冲液,流动相B为pH 7.20的醋酸盐缓冲液-乙腈-甲醇,两相均为较高浓度的盐溶液,对色谱柱、液相泵的使用寿命影响较大;②流动相B为盐溶液与有机溶剂的混合溶液,该混合溶液为不稳定体系,在分析过程中可能会有盐析出,影响氨基酸的分离;③该系统对流动相pH非常敏感,要求我们配制A、B相的pH误差范围应为7.20±0.05,如有误差则会影响各氨基酸的分离度。
改进后的色谱条件A相为一定浓度的磷酸盐溶液,B相则为不含盐的乙腈、甲醇、水的混合液,降低了对色谱柱、液相泵的损耗。且该系统对流动相的pH敏感性较低,pH的误差对氨基酸分离的影响不大,也大大提高了流动相配制的便捷性。
3.4盐酸对于色谱柱的损耗
色氨酸、丙氨酸、缬氨酸、甲硫氨酸、异亮氨酸、苯丙氨酸、亮氨酸和脯氨酸为疏水氨基酸,均难溶于水,在原有操作规程中我们采用0.04 mol/L盐酸溶液作为溶剂配制对照品溶液。在大量的试验过程中,我们发现盐酸对于色谱柱存在损耗,使得色谱柱柱效下降,寿命缩短。故我们改进了对照品溶液的配制方式。先将以上氨基酸配制为高浓度的对照品贮备液(配制浓度为测试浓度的5倍,溶剂为0.04 mol/L盐酸溶液),使其完全溶解,再使用双蒸水稀释至合适浓度,这种配制方式大大降低了对照品溶液中盐酸的浓度,更好的保护了色谱柱。
在水解氨基酸样品前处理时,我们需将脑蛋白水解物原料用浓盐酸水解后制备供试样品溶液。在处理过程中我们将酸水解后的样品水浴蒸干,再使用双蒸水溶解并稀释。这一过程的目的同样是将供试品中的盐酸尽可能去除,起到保护色谱柱的作用。
4 结论
经方法学验证,本研究所建立的肽含量测定方法操作简便、专属性强,能准确测定脑蛋白水解物原料中肽的含量。
[1] 李婷, 刘力, 徐德生. 氨基酸柱前衍生化在动物药质量控制中的应用[J]. 中国药业, 2014, 23(2): 16-19.
[2] 刘晓剑. Accq.Tag柱前衍生法分析蜂胶中的氨基酸含量[J]. 检验检疫科学, 2000, 10(5): 47-48.
[3] 庞新安, 万英. AccQ-Tag柱前衍生反相高效液相色谱法测定果品中氨基酸含量[J]. 分析仪器, 2007, 4: 32-35.
[4] 陈宇堃, 梁蔚阳, 薛巧如, 等. AccQ-Tag法测定复方氨基酸注射液(18AA—V)中17种氨基酸的含量[J]. 广东药学院学报, 2008, 24(2): 243-246.
[5] 牟德海. OPA柱前衍生反相高效液相色谱法测定氨基酸含量[J]. 色谱, 1997, 15(4): 319-321.
[6] 叶英. OPA-FMOC柱前衍生反相液相色谱法测定氨基酸含量[J]. 食品科学, 2002, 23(9): 91-93.
[7] 崔淑芬, 许柏球, 王小如. 柱前衍生RP-HPLC法测定泽泻中氨基酸的含量[J]. 中草药, 2004, 35(8): 867-869.
[8] 初虹, 徐龙芳, 盛伟红, 等. 高效液相色谱法测定胸腺肽a1的氨基酸组成[C]. 中国药学会、河北省人民政府,2 0 0 8 中 国 药 学 会 学 术 年 会 暨 第 八 届 中 国 药 师 周 论 文 集.
Optimization of a method for the determination of peptide content in cerebroprotein hydrolysate
ZHANG Sumin, TANG Xiaoshu*, ZHOU Yuanyuan
(Shanghai Greatwall Pharmaceutical CO. LTD., Shanghai 201206, China)
Objective: To optimize a method for the determination of peptide content in cerebroprotein hydrolysate. Methods: HPLC was performed on an Agilent Zorbax Eclipse-AAA column(4.6 mm×150 mm, 5 mm)with mobile phase A: 0.04 mol/L phosphate buffer (pH 7.8) and mobile phase B: acetonitrile-methanol-water (45:45:10) at UV detection wavelength of 338 nm and 262 nm, flow rate of 1.5 ml/min (gradient elution) and column temperature of 40 ℃. Results: The standard curve of every amino acid was linear over the range of 15.0~120.0 mg/ml (r>0.999)with average recovery 98.1%~103.6%. Conclusion: This method is simple, accurate, sensitive and reproducible, and can be applied in the determination of peptide content in cerebroprotein hydrolysate.
cerebroprotein hydrolysate; determination of peptide content; amino acids; HPLC
R927.2; O657.72
A
1006-1533(2015)13-0074-06
汤晓枢(1979-), 男, 工程师, 从事化学与生化药品研发工作。E-mail: tangxiaoshu_1979@126. com
2015-03-03)



