高碧霞,李明喜,刘雪娇,蔡建芳,樊晓红,杨啸林,李雪梅,李学旺
中国医学科学院 北京协和医学院 北京协和医院 1肾内科 2转化医学中心,北京 100730 3中国医学科学院 北京协和医学院 基础医学研究所生物医学工程系,北京 100005
代谢综合征 (metabolic syndrome,MS)是一组以胰岛素抵抗为病理生理基础的代谢紊乱症候群[1]。研究显示MS是慢性肾脏疾病 (chronic kidney disease,CKD)独立危险因素,但目前尚缺乏MS早期肾损害生物标志物及诊断方法的研究[1]。蛋白质组学技术在筛选疾病生物标志物方面具有广阔的发展前景[2]。纳米磁珠联合基质辅助激光解析电离飞行时间质谱 (matrix-assisted laser desorption ionization time-of-flight mass spectrometry,MALDI-TOF-MS)技术是近年发展较快的一项临床蛋白质组学技术,有研究应用该技术探讨肾小球疾病尿液多肽谱,发现不同肾小球疾病呈现各自独特的尿液多肽表达谱,提示该技术可作为寻找肾脏疾病尿液潜在标志物的有效手段[3]。本研究采用弱阳离子交换磁珠 (magnetic bead-based weak cation exchange chromatography,MB-WCX)联合MALDI-TOF-MS建立MS早期肾损害尿液蛋白谱并寻找疾病潜在尿液标志物。
对象和方法
对象 来源于2008至2009年北京平谷地区“MS肾脏损害”流行病学研究[4],入选者留取8h过夜尿液标本,女性留尿避开月经期,标本收集当日避免剧烈运动和饮酒。采样前均签署知情同意书。
方法 入选者进行问卷调查、体格检查和实验室检查,同文献 [4]。入选和排除标准:入选者分为MS无肾脏损害组和MS早期肾脏损害组。MS按照美国国家胆固醇教育计划的成人治疗专家组Ⅲ诊断标准[1]。MS早期肾损害符合以下两个条件[5]:(1)20μg/min≤尿白蛋白排泄率 < 200μg/min;(2)由简化“肾脏病膳食改良试验”公式[6]计算的肾小球滤过率 (estimated glomerular filtration rate,eGFR)≥60 ml/(min·1.73 m2)。排除标准:(1)高血压3级:两次测量血压平均值收缩压≥180和 (或)舒张压≥110 mmHg(1 mmHg=0.133 kPa);(2)血尿及脓尿:尿沉渣镜检白细胞≥5个/高倍镜视野或红细胞≥3个/高倍镜视野;(3)其他慢性肾脏疾病、泌尿系结石、慢性肝炎及近期泌尿系统感染病史。
MB-WCX富集尿液蛋白 采用MB-WCX试剂盒(德国Bruker Daltonics公司)进行尿液蛋白的分离和富集,具体步骤参考文献 [7]。
MALDI-TOF-MS建立质谱图 应用UltrafleX-tremeTMⅢ MALDI-TOF/TOF质谱仪 (德国Bruker Daltonics公司)建立尿液蛋白谱图,步骤如下:(1)点靶:参考文献 [7];(2)质谱数据采集:采用正离子线性模式,参数设置如下:第一离子源20.0 kV,第二离子源18.5 kV,以1000 Hz氮激光照射,激光能量35% ~45%,检测范围为质荷比1000~15000。每个标本点3个靶点,同一靶点多点采集共累积500次建立质谱图。
实验重复性评估和质量控制 实验重复性评估方法见参考文献 [6]。数据采集前每6个靶点取1个标准品 (德国Bruker Daltonics公司)进行质谱仪质量校准,分子质量的误差范围<0.1%。
生物信息学软件处理
谱图处理:ClinProTools(CRT)2.1软件进行数据图谱处理,包括基线平滑、衰减、过滤掉信噪比<5的峰等,对数据进行校正和归一化处理。
差异蛋白峰的筛选及模型构建:有两种方法。(1)统计学方法联合遗传算法 (genetic algorithm,GA):以峰面积作为质荷比蛋白峰的量化指标,Wilcoxon检验进行组间比较,P<0.05为差异具有统计学意义。通过CRT软件内置的GA算法对上述差异蛋白峰进行筛选并构建诊断模型。采用10倍交叉验证对模型进行评估。(2)随机森林 (random forest,RF)联合支持向量机 (support vector machine,SVM):质谱数据同谱图处理,通过RF筛选差异蛋白峰,以蛋白峰“mean decrease in accuracy”作为评估每个蛋白峰在分类中重要性的标准,重要性>0.005的峰作为差异蛋白峰;利用SVM(LibSVM2.88)对差异蛋白峰构建诊断模型,核函数采用径向基核函数,通过网格搜索和10倍交叉验证优化模型,确定SVM参数C=2.65,gamma=2-14.5时模型分类性能最佳。绘制受试者工作特征曲线并计算曲线下面积。
统计学处理 采用SPSS 11.5统计软件,计量资料组间比较采用t检验,计数资料组间比较采用χ2检验。P<0.05为差异具有统计学意义。
结 果
平均谱图的建立 入选者包括MS无肾损害患者54例和 MS早期肾损害患者46例,性别、年龄、eGFR两组比较差异无统计学意义,尿白蛋白排泄率在MS早期肾损害组显着高于MS无肾脏损害组 (P<0.05)(表1)。实验重复性评估通过计算变异系数为7.7%~23.0%。应用CRT软件进行谱图处理,建立两组样本尿液平均蛋白谱图 (图1)。
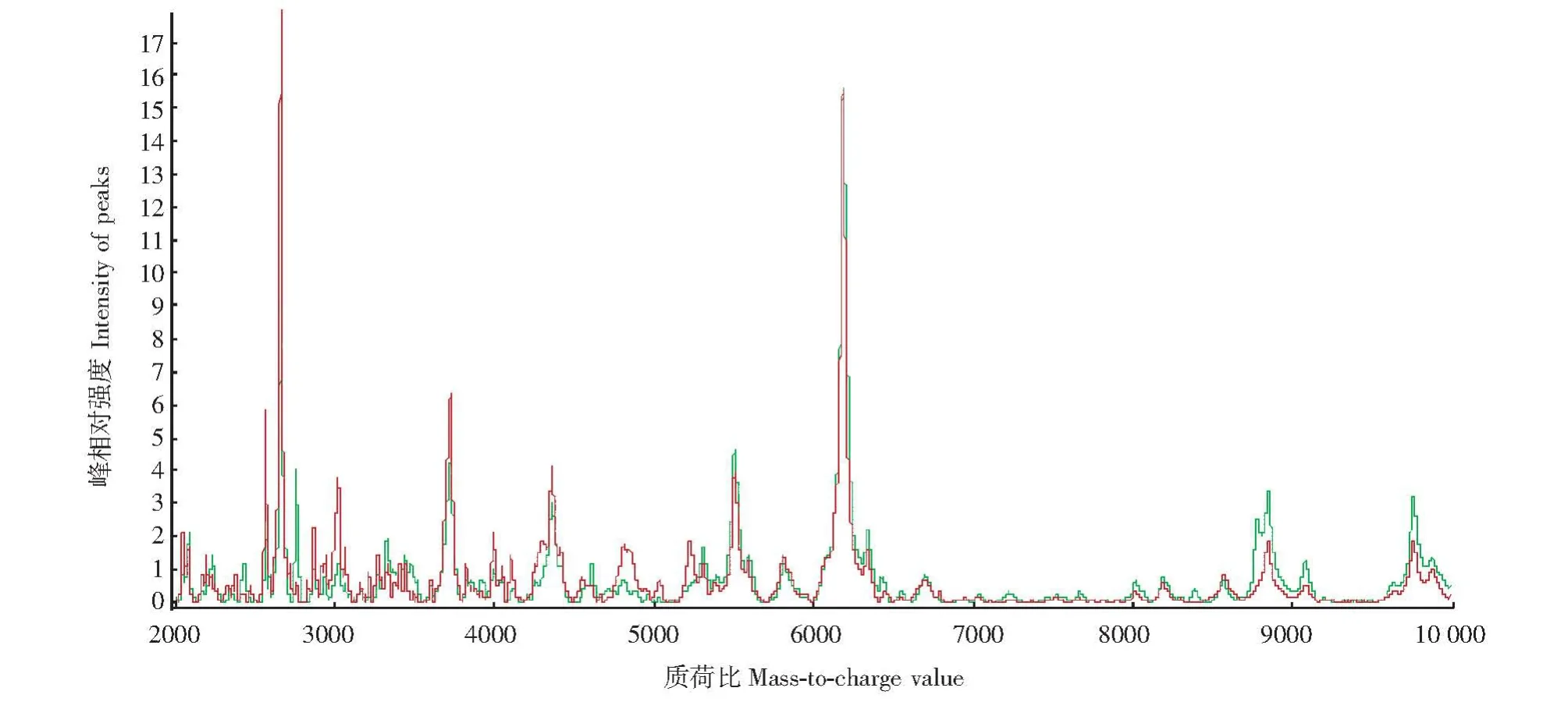
图1 MS无肾脏损害组 (红色)和MS早期肾脏损害组 (绿色)的平均尿液蛋白图谱Fig 1 Average urinary protein spectra of MS without renal injury(red)and MS with early renal injury(green)
差异蛋白峰的筛选 应用Wilcoxon检验分析两组平均尿液蛋白谱图,在相对分子质量1000~15000内,38个蛋白峰表达差异具有统计学意义 (P<0.05),其中20个蛋白峰在MS早期肾损害组高表达;应用 RF算法显示“mean decrease in accuracy”重要性>0.005的蛋白峰14个,12个蛋白峰在MS早期肾损害组表达上调,这12个蛋白峰中有11个亦是Wilcoxon检验发现的差异蛋白峰 (表2)。
诊断模型的建立和评估 GA算法筛选5个蛋白峰构建诊断模型,其中3个蛋白峰 (质荷比2756.98、9077.04和10054.26)在MS早期肾损害组表达上调。该模型对MS早期肾损害诊断敏感性为82.6%、特异性为84.3%、准确性为83.5%。RF算法筛选14个差异蛋白峰应用于SVM算法构建诊断模型 (表2),此模型对MS早期肾损害诊断敏感性为89.2%、特异性为81.1%、准确性为85.5%。对RF联合SVM构建模型绘制受试者工作特征曲线,曲线下面积为0.91(图2)。两个模型中共有的蛋白峰包括质荷比2756.98、3019.11、9077.04和10054.26,其中质荷比2756.98、9077.04和10054.26在MS早期肾损害患者尿液中高表达 (表2、3)。
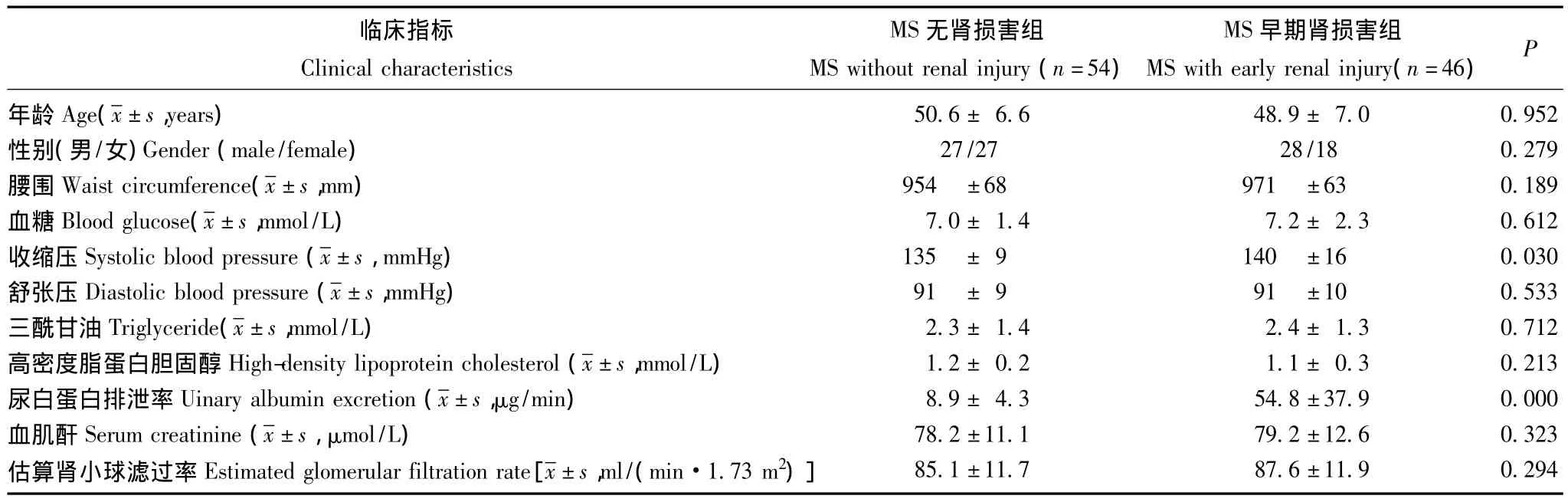
表1 MS无肾损害和MS早期肾损害患者临床资料Table 1 Clinical data of metabolic syndrome patients without/with renal injury
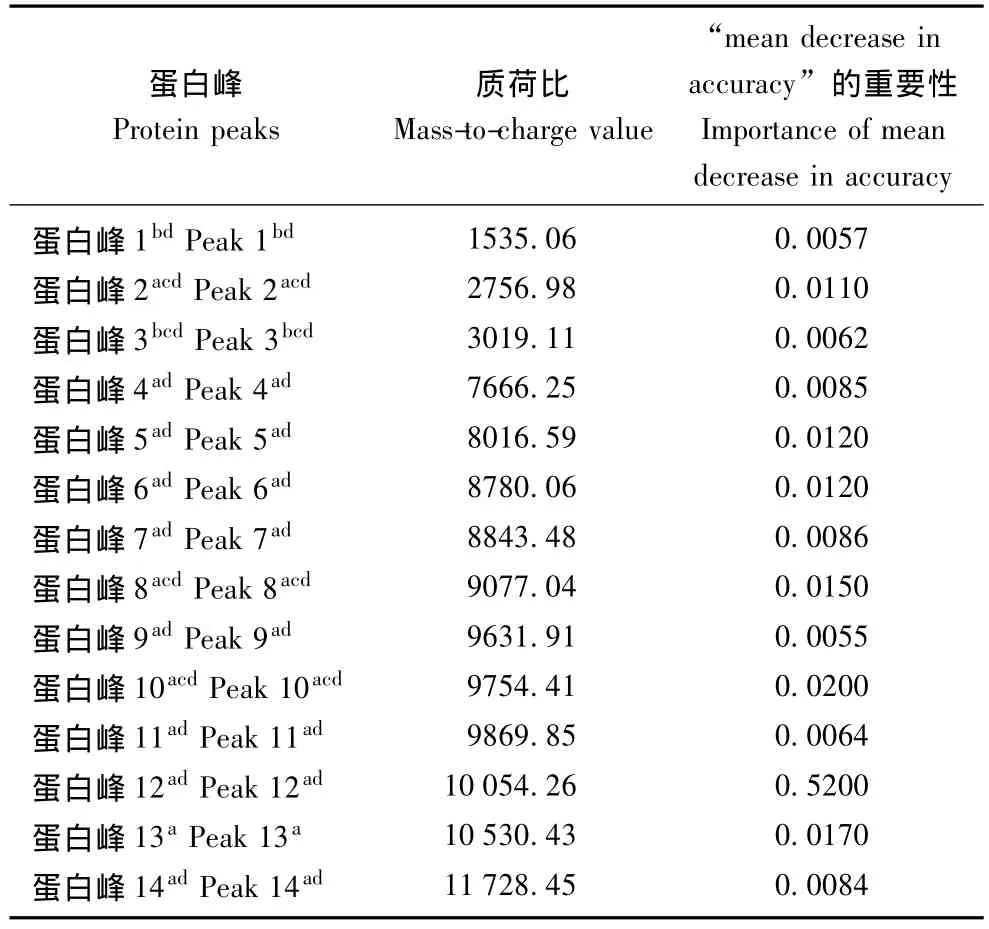
表2 RF-SVM算法构建诊断模型中的蛋白峰Table 2 Depiction of protein peaks in RF-SVM-based diagnostic model
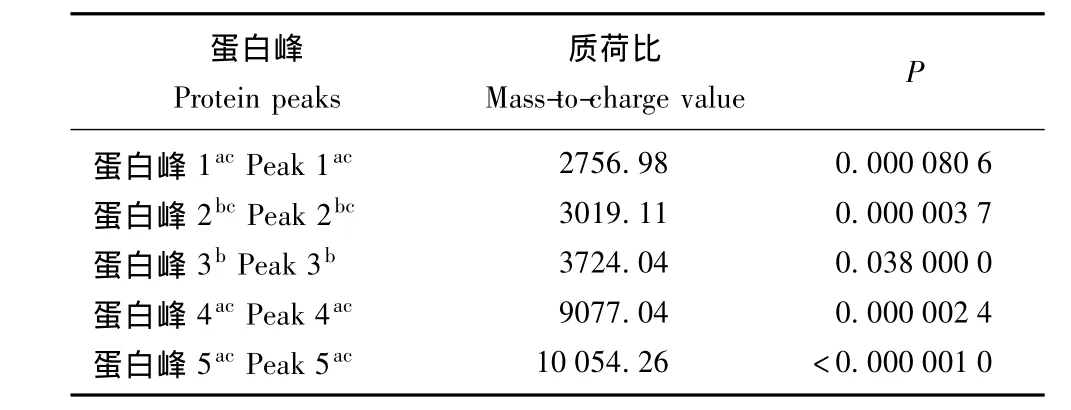
表3 GA构建诊断模型中的蛋白峰Table 3 Depiction of protein peaks in GA-based model
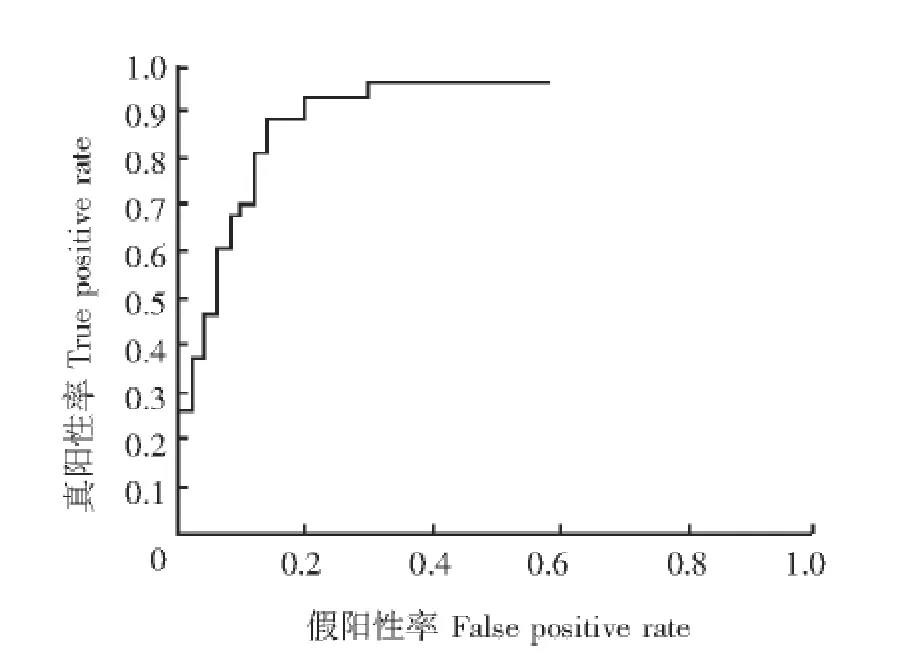
图2 SVM分类器构建诊断模型的受试者工作特征曲线Fig 2 Receiver operating characteristic curve of SVM-based diagnostic model
讨 论
随着生活方式的改变,MS发病率逐年升高[1]。研究显示MS是CKD独立的危险因素,对MS肾脏损害早期诊断和干预对改善相关肾脏疾病的长期预后具有重要的意义[1]。目前微量白蛋白尿和eGFR下降是MS早期肾脏损害的主要临床指标,有些糖尿病患者出现微量白蛋白尿并不进展为临床蛋白尿,而eGFR下降是所有CKD患者病情进展的表现。针对MS早期肾脏损害迄今尚无较好的临床监测指标。
临床蛋白质组学在疾病生物标志物研究领域应用广泛。尿液是最容易得到的体液,主要反映肾脏、膀胱及全身其他系统的生理和病理状态,其收集方法获取简单无创,蛋白质/多肽含量丰富,是较好的临床蛋白质组学研究资源,特别适合肾脏疾病生物标志物的研究[8]。尿液蛋白质组学常用的技术包括二维凝胶电泳、液相色谱联合串联质谱 (liquid chromatography coupled to tandem mass spectrometry,LCMS/MS)以及表面增强激光解吸电离飞行时间质谱(surface-enhanced laser desorption/ionization time of flight mass spectrometry, SELDI-TOF-MS) 技 术[9]。SELDI-TOF-MS技术通过载有不同配基的芯片捕获小分子蛋白/多肽,灵敏度高达飞摩尔,作为一项高通量的蛋白质组学技术一定程度上弥补了二维凝胶电泳和LC-MS/MS的不足,但该技术重复性欠佳,不能直接鉴定差异蛋白[10]。2004年出现的Clinprot系统应用球形纳米磁珠富集蛋白,和蛋白芯片相比,由于结合表面积增大,捕获的小分子蛋白/多肽的种类更多,敏感性和准确性更高;联合高灵敏度的MALDI-TOF质谱仪,实验重复性得到了提高,在临床大样本研究中更具优势[11];可联合LC-MS/MS鉴定高丰度的小分子蛋白/多肽[12]。
在尿液蛋白质组学研究中,样本的收集和处理是影响实验结果的关键。Fiedler等[7]通过研究尿液收集、储存等方法的不同对磁珠富集尿液中蛋白及多肽的影响,建立了磁珠分离尿液蛋白的标准化流程。本研究在尿液标本的收集和处理过程中借鉴上述标准化流程,规范从流调样本采集、运送、处理以及存储各个环节,最大程度上避免人为因素对标本中蛋白含量的影响;每个样本冻融1次;采用标准品进行相对分子质量校正,保证校正的平均相对分子质量偏差<0.01%;在数据采集方面,每例制备好的尿液标本在靶上重复3个点,对同一靶点的不同结晶点进行多点采集。得到的变异系数较好,保证了实验的稳定性和重复性。
本研究采用WCX磁珠联合UltrafleXtremeTMⅢMALDI-TOF/TOF质谱仪建立了MS无肾损害和MS早期肾损害患者尿液蛋白谱图。通过CRT软件内置的Wilcoxon检验筛选出20个蛋白峰在 MS早期肾脏损害患者尿液中高表达;应用RF算法筛选出12个蛋白峰在MS早期肾脏损害组尿液中高表达,其中11个蛋白峰也是Wilcoxon检验结果中的差异峰,提示这11个差异蛋白峰可能作为MS早期肾损害尿液诊断标记物的候选蛋白。目前尚无应用尿液蛋白质组学技术探索MS早期肾脏损害诊断标记物的研究,Gianazza等[12]采用C8磁珠联合MALDI-TOF MS技术对糖尿病肾病和正常人血清蛋白质组进行了研究,联合LC-MS/MS鉴定纤维蛋白肽A在糖尿病肾病患者血清高表达。本研究室下一步通过磁珠富集目标蛋白,应用LC-MS/MS对候选蛋白进行序列鉴定。
机器学习分类方法已广泛应用于蛋白质组学中质谱数据的模型构建,模型通过多个特征峰的联合检测代替单一特征峰,提高了疾病诊断的敏感性和特异性。在多种机器学习分类方法中SVM和RF对质谱数据的分类能力最好,且RF在数据特征点选择方面具有独特的优势[13]。本研究在差异峰的选择和诊断模型的构建中,除了采用CRT软件内置的GA算法,还将RF联合SVM算法应用Clinprot系统的质谱数据分析。采用多种机器分类方法构建的两个模型共同包含了质荷比2756.98、3019.11、9077.04和10054.26个蛋白峰,结果具有较好的一致性,交叉验证评估两个模型对MS早期肾损害诊断的敏感性、特异性和准确性均较好,下一步可增加临床样本量对诊断模型加以验证。
综上,本研究应用WCX磁珠分离结合MALDITOF-MS分析MS早期肾损害尿液蛋白谱图,采用多种机器学习分类方法筛选出MS早期肾脏损害的尿液差异蛋白峰,并建立了具有较好识别率的诊断模型。下一步将进一步进行差异蛋白峰的序列鉴定和验证。
[1]Agrawal V,Shah A,Rice C,et al.Impact of treating the metabolic syndrome on chronic kidney disease [J].Nat Rev Nephrol,2009,5(9):520-528.
[2]Mischak H,Apweiler R,Banks RE,et al.Clinical proteomics:a need to define the field and to begin to set adequate standards [J].Proteomics Clin Appl,2007,1(2):148-156.
[3]吴杰,李燕,陈香美,等.磁珠分离结合生物质谱分析肾小球疾病患者尿液多肽谱 [J].中华肾脏病杂志,2009,25(8):596-600.
[4]樊晓红,蔡建芳,李学旺,等.中国汉族人以尿白蛋白肌酐比值诊断微量白蛋白尿的界值研究[J].中华肾脏病杂志,2010,26(11):807-811.
[5]Chen B,Yang D,Chen YU,et al.The prevalence of microalbuminuria and its relationships with the components of metabolic syndrome in the general population of China [J].Clinica Chimica Acta,2010,411(9-10):705-709.
[6]Levey AS,Coresh J,Greene T,et al.Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate[J].Ann Intern Med,2006,145(4):247-254.
[7]Fiedler GM,Baumann S,Leichtle A,et al.Standardized peptidome profiling of human urine by magnetic bead separation and matrix-assisted laser desorption/ionization time-offlight mass spectrometry[J].Clin Chem,2007,53(3):421-428.
[8]Bramham K,Mistry HD,Poston L,et al.The non-invasive biopsy-will urinary proteomics make the renal tissue biopsy redundant[J]?Q J Med,2009,102(8):523-538.
[9]Welberry Smith MP,Banks RE,Wood SL,et al.Application of proteomic analysis to the study of renal diseases[J].Nat Rev Nephrol,2009,5(12):5701-5712.
[10]Poon TC.Opportunities and limitations of SELDI-TOF-MS in biomedical research:practical advices[J].Expert Rev Proteomics,2007,4(1):51-65.
[11]Zhang X,Leung SM,Morris CR,et al.Evaluation of a no-vel,integrated approach using functionalized magnetic beads,bench-top MALDI-TOF-MS with prestructured sample supports,and pattern recognition software for profiling potential biomarkers in human plasma [J].J Biomol Tech,2004,15(3):167-175.
[12]Gianazza E,Mainini V,Castoldi G,et al.Different expression of fibrinopeptide A and related fragments in serum of type 1 diabetic patients with nephropathy[J].J Proteomics,2010,73(3):593-601.
[13]Datta S,Pihur V.Feature selection and machine learning with mass spectrometry data[M]//Matthiesen R.Bioinformatics methods in clinical research,methods in molecular biology.New York:Humana Press,2010:205-229.



