孙 邈,崔 琳,刘卫红,高 原,沈 思,朱明军,王幼平
1河南中医学院第一附属医院中心实验室及心脏中心,郑州 450000
2河南中医学院中医内科学,郑州 450008
高血压,尤其是盐敏感性高血压是造成终末期肾脏病的主要疾病之一[1-3]。大量研究表明以单核/巨噬细胞浸润为主的炎症反应在盐敏感性高血压所引起的肾脏损害中发挥重要作用[4]。在病理状态下,白细胞与趋化因子相互作用促使白细胞渗出并向损伤和炎症部位聚集,从而形成炎症反应。单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)属于C-C趋化因子家族。MCP-1与单核/巨噬细胞表面的C-C趋化因子受体2(C-C chemokine receptor 2,CCR2)相互作用,从而诱发以单核/巨噬细胞浸润为主的炎症反应[5]。实验研究显示,通过使用药物阻断CCR2受体可明显抑制多种疾病引起的肾脏损害,其中包括糖尿病引起的肾脏损害[6]。本研究拟在醋酸脱氧皮质酮 (desoxycorticosterone acetate,DOCA)-盐高血压动物模型上观察CCR2受体阻断剂对肾脏功能和形态学的影响,以阐明CCR2受体对盐敏感性高血压造成的肾脏损害的影响。
材料和方法
实验动物及试剂 清洁级雄性C57BL/6小鼠(周龄8~9周)购于北京维通利华实验动物技术有限公司,许可证号:SCXK(京)2011-0011。CCR2受体阻断剂 RS504393(Tocris Bioscience,USA),DOCA 片 (Innovative Research of America,USA),ELISA尿白蛋白试剂盒 (Exocell,USA);改良的Jaffe肌酐测定试剂盒 (BioAssay System,USA);尿8-异构前列腺素 (8-isoprostane)试剂盒 (Ann Arbor,Cayman Chemical公司);大鼠抗小鼠F4/80单克隆抗体 (1∶200,Serotec,UK),大鼠 IgG(1∶400,Vector Laboratories,USA)。
造模及分组 C57BL/6小鼠经过1周适应性饲养后,分为DOCA-盐高血压模型组、DOCA-盐高血压RS504393组和对照组。DOCA-盐高血压小鼠根据本实验室已建立的实验方法[4]制备。方法如下:经皮下注射混合麻醉剂 (氯胺酮80 mg/kg+甲苯噻嗪4 mg/kg)麻醉小鼠,常规切除左侧肾脏,于小鼠颈部皮下植入DOCA片 (Innovative Research of America,USA),术后给予含1%NaCl和0.2%KCl的饮用水。DOCA-盐高血压模型组给予RS504393的溶媒;DOCA-盐高血压RS504393组给予特异性的CCR2受体阻断剂RS504393(RS504393溶于含有5%二甲基亚砜的生理盐水,皮下注射,2 mg/kg,每日2次,持续4周);对照组小鼠仅摘除左侧肾脏,但不植入DOCA片,给予正常饮用水。术后所有小鼠连续3d肌肉注射青霉素钠盐。实验共观察4周。
动脉收缩压测定 分别于术前及术后各周,根据Tail-cuff体积描记法原理利用BP-2000型血压测定仪 (Visitech System,USA)测定清醒小鼠尾动脉收缩压。测定时间为上午10:00~12:00。小鼠在30℃烘箱内安静加热10 min后,固定器固定,待小鼠稳定后连续测定尾动脉收缩压5次,取其平均值作为该小鼠动脉收缩压。
尿样及血浆分析 于术后第4周结束前2 d,将小鼠放入代谢笼,适应后收集24 h尿液,称体重。皮下注射混合麻醉剂麻醉小鼠,经腹主动脉采血、离心,收取血浆,保存于-80℃。尿白蛋白用ELISA试剂盒(Exocell,USA)测定;尿及血浆中肌酐浓度用改良的Jaffe肌酐测定试剂盒测定 (BioAssay System,USA);尿8-异构前列腺素用Cayman Chemical公司 (Ann Arbor,USA)生产的特异性试剂盒测定。
肾脏病理组织学分析 摘除右肾、称重,置于4%多聚甲醛固定,常规石蜡包埋。对4 μm厚石蜡包埋组织切片进行过碘酸-雪夫 (PAS)染色和Masson三色染色。由1位不了解实验情况但具有病理学知识的人员,根据文献 [4],通过以上两种染色分别对肾小球纤维样硬化和肾小管间质损伤程度进行分析。根据肾小球硬化范围、肾小管扩张、肥厚、间质纤维化及炎症细胞浸润程度进行半定量分析。
免疫组织化学染色分析 采用卵白素-生物素复合物技术对单核/巨噬细胞标记物F4/80进行染色。对石蜡包埋组织切片常规处理后,加入大鼠抗小鼠F4/80单克隆抗体,4℃下孵育12 h,冲洗,然后加入生物素标记的抗大鼠IgG(1∶400),室温下孵育1 h后加卵白素-生物素复合物,室温孵育45 min,最后加入二氨基联苯胺显色液,显微镜下观察控制染色。实验同时采用正常大鼠血清或去除第一抗体的方法作为阴性对照。根据文献 [4],每张切片随机选取15个视野,在高倍镜 (×400)下对F4/80阳性细胞计数,最后取其平均值代表单核/巨噬细胞数量。
结 果
各组小鼠血压 术前各实验组之间基础血压差异无统计学意义。术后对照组小鼠血压平稳,无明显增加,而术后第1周模型组和RS504393组小鼠血压较对照组明显升高 (P<0.05),并且于术后第3、4周达高峰。然而,模型组和RS504393组小鼠血压之间差异无统计学意义 (图1)。
尿白蛋白排泄量 术前及术后观察期间,各实验组小鼠之间体重差异无统计学意义。与对照组相比,模型组小鼠肾脏/体重比值和24 h尿白蛋白排泄量显着增加 (P<0.05),而肌酐清除率明显下降(P<0.05),RS504393处理可显着抑制以上变化,使其恢复正常 (表1)。
尿8-异构前列腺素24 h排泄量 尿8-异构前列腺素24 h排泄量作为反映肾脏氧化应激反应的指标,术后模型组小鼠较对照组小鼠明显增加 (P<0.05),RS504393可明显抑制其增加 (图2)。
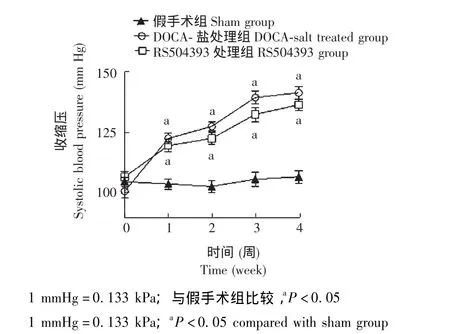
图1 各组小鼠收缩压比较Fig 1 Changes in systolic blood pressure in all groups
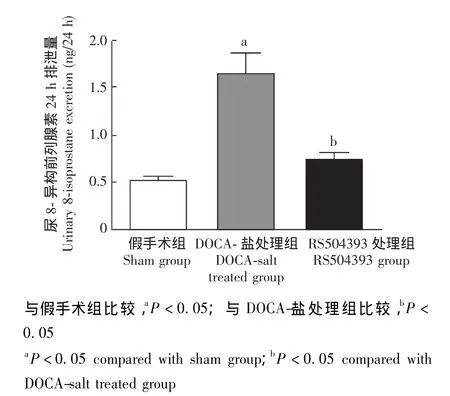
图2 各组小鼠尿8-异构前列腺素24 h排泄量Fig 2 Changes in urinary 8-isoprostane excretion over 24-hour period in all groups
表1 各组小鼠体重及肾脏相关参数的变化 ()Table 1 Changes in body weight and renal parameters in all groups()
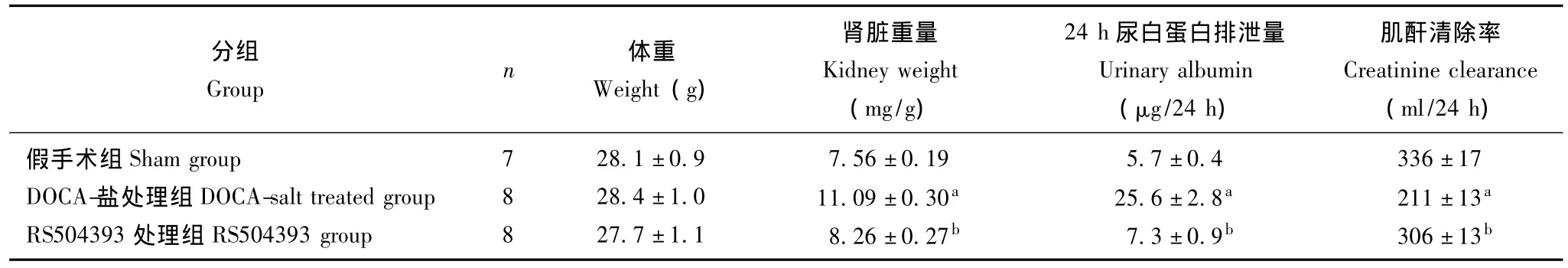
表1 各组小鼠体重及肾脏相关参数的变化 ()Table 1 Changes in body weight and renal parameters in all groups()
与假手术组比较,aP<0.05;与DOCA-盐处理组比较,bP<0.05aP<0.05 compared with sham group;bP<0.05,compared with DOCA-salt treated group
分组Group n 体重Weight(g)肾脏重量Kidney weight(mg/g)24 h尿白蛋白排泄量Urinary albumin(μg/24 h)肌酐清除率Creatinine clearance(ml/24 h)假手术组Sham group 7 28.1±0.9 7.56±0.19 5.7±0.4 336±17 DOCA-盐处理组 DOCA-salt treated group 8 28.4±1.0 11.09±0.30a 25.6±2.8a 211±13a RS504393处理组 RS504393 group 8 27.7±1.1 8.26±0.27b 7.3±0.9b 306±13b
肾脏病理组织学 模型组小鼠肾小球可见明显的纤维样硬化,其肾小球纤维样硬化指数较对照组显着升高,RS504393处理可抑制其升高,防止盐敏感性高血压诱发的肾小球纤维样硬化 (图3)。模型组小鼠肾小管间质损伤程度较对照组明显加重,主要表现为肾小管扩张与肥厚、间质纤维化及炎性细胞浸润等,RS504393处理可抑制上述变化,阻止盐敏感性高血压诱发的肾小管间质损伤 (图4)。
免疫组织化学染色 F4/80阳性的单核/巨噬细胞数量在模型组小鼠肾脏较对照组小鼠显着增加,RS504393处理可明显抑制F4/80阳性的单核/巨噬细胞数量在肾内升高,从而阻止盐敏感性高血压诱发的以单核/巨噬细胞浸润为主的肾内炎性反应 (图5)。
讨 论
本研究显示通过皮下包埋DOCA和饮用盐水诱发的盐敏感性高血压造成的肾脏损害,其主要表现为尿白蛋白增加、肌酐清除率下降、尿8-异构前列腺素排泄增加、肾小球纤维样硬化、肾小管间质损伤,其中包括肾小管扩张和肾间质纤维化以及肾间质单核/巨噬细胞浸润等。所有这些肾脏功能及形态学的变化均被CCR2受体阻断剂所抑制。表明CCR2受体介导盐敏感性高血压诱导的肾脏损害,其机制可能通过CCR2受体介导的单核/巨噬细胞浸润。
在高血压过程中,血压升高是造成肾脏损害的主要机制之一[7]。本研究通过使用鼠尾动脉法确定小鼠尾动脉收缩压,结果显示小鼠通过皮下包埋DOCA和饮用盐水可诱发盐敏感性高血压,阻断CCR2受体可抑制和改善盐敏感性高血压造成的肾脏损害,但是,CCR2受体阻断剂并不影响皮下包埋DOCA和饮用盐水诱发的盐敏感性高血压,两组小鼠的血压之间差异无统计学意义。因此,在盐敏感性高血压造成的肾脏损害过程中,CCR2受体阻断剂诱导的肾脏保护作用结果主要来源于该阻断剂对CCR2受体介导的信号转导通路的影响而非对血压的影响。表明在盐敏感性高血压过程中CCR2受体阻断剂介导的肾脏保护作用是独立于血压因素的,并非通过降低血压而实现其肾脏保护作用。
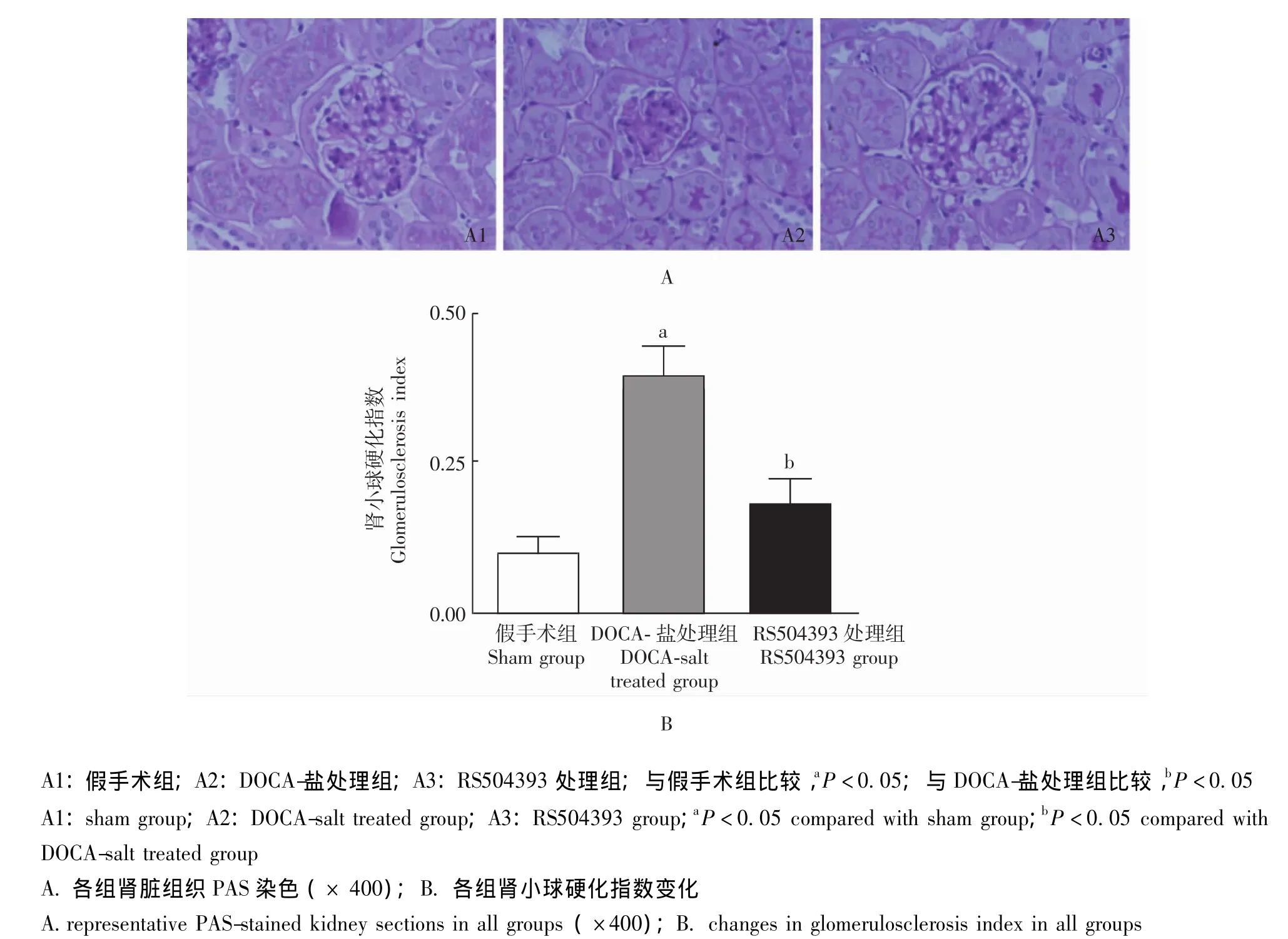
图3 各组小鼠肾小球纤维样硬化程度Fig 3 Changes in glomerulosclerosis in all groups
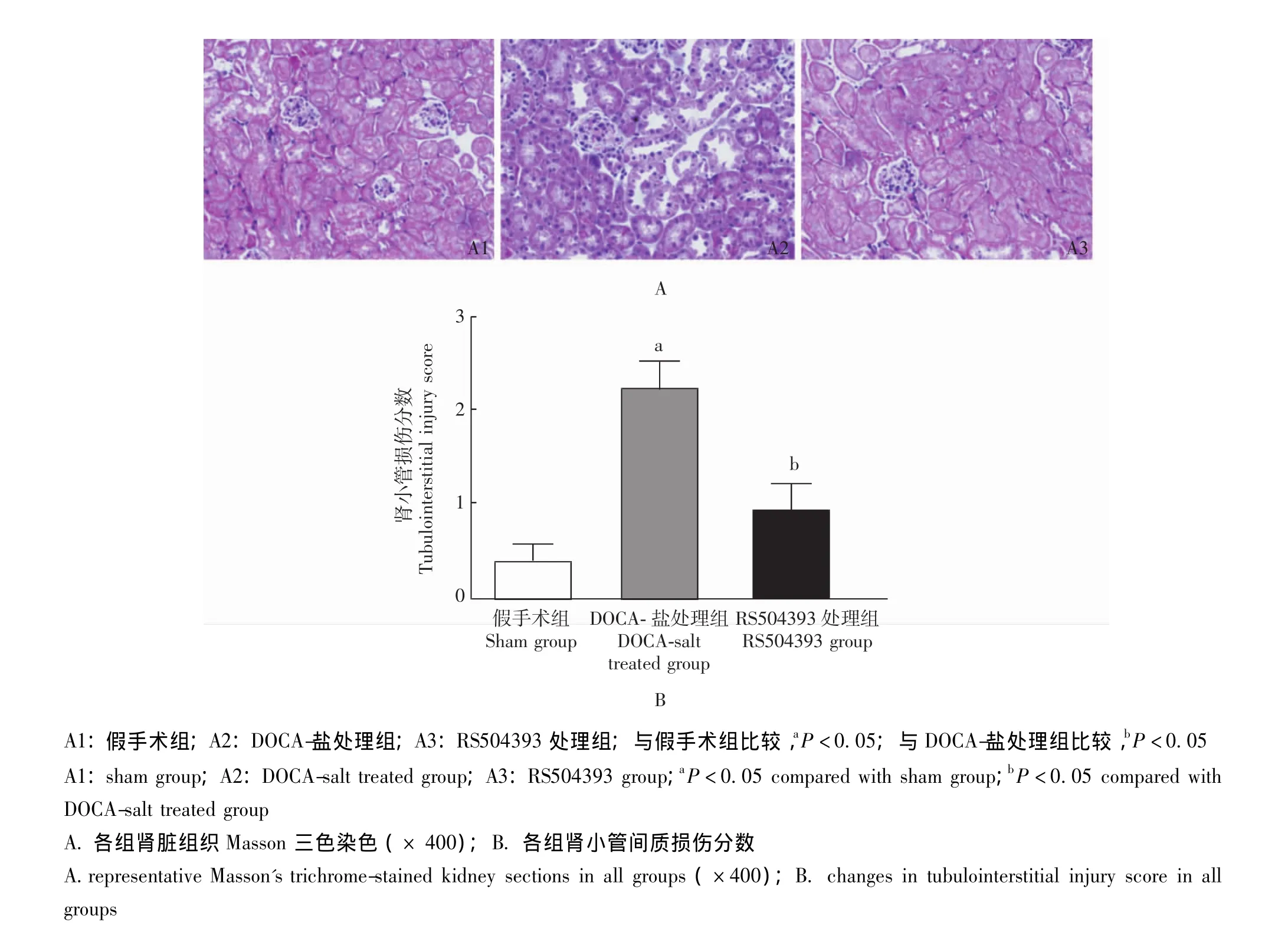
图4 各组小鼠肾小管间质损伤程度Fig 4 Changes in tubulointerstitial injury in all groups
尿白蛋白是反映终末期肾脏疾病严重程度的重要指标。本研究通过皮下包埋DOCA和饮用盐水诱发的盐敏感性高血压在小鼠上造成明显的白蛋白尿。需要指出的是尿白蛋白的明显增加却仅伴随中等程度的肾小球损害,即尿白蛋白的变化并不平行于光学显微镜观测到的肾小球的损害。有研究显示在光学显微镜观测到肾脏损害之前尿白蛋白就已经有明显增加[8-9],其机制在于导致白蛋白尿的肾脏超微结构的改变发生在光学显微镜可观测到的改变之前,表明超微结构的改变,包括肾小球基质膜的增厚和足细胞的退化,都先于光学显微镜下组织形态学的改变[10]。大量研究证据表明肾小球足细胞损伤与白蛋白尿密切相关[11]。CCR2受体对盐敏感性高血压造成的肾脏超微结构损害的影响有待于进一步的研究。
尽管盐敏感性高血压造成肾脏损害的具体机制尚未完全阐明,但有研究已经证明炎性反应,尤其是以单核/巨噬细胞浸润为主的炎性反应在此过程中发挥重要作用[4,12]。本研究在通过皮下包埋DOCA和饮用盐水诱发的盐敏感性高血压过程中,肾脏损害伴随有明显的单核/巨噬细胞浸润[4,13],提示单核/巨噬细胞可能在盐敏感性高血压造成的肾脏损害中发挥重要作用。血中的白细胞包括单核细胞在其渗出、移动和分化过程中涉及到多种环节和因素,在此过程中趋化因子发挥重要作用。在各种趋化因子中,MCP-1对单核/巨噬细胞的渗出和移动发挥重要的调节作用。MCP-1与单核/巨噬细胞表面的CCR2受体相互作用促使单核/巨噬细胞的渗出、移动和分化,从而形成以单核/巨噬细胞浸润为主的炎症反应[5]。本研究CCR2受体阻断剂抑制盐敏感性高血压造成的肾脏损害,该保护作用同时伴随有单核/巨噬细胞浸润的下降。因此,推测在盐敏感性高血压过程中,CCR2受体阻断剂介导的肾脏保护作用可能是通过抑制MCP-1与CCR2受体之间的相互作用而减少单核/巨噬细胞的浸润,以达到其肾脏保护作用。
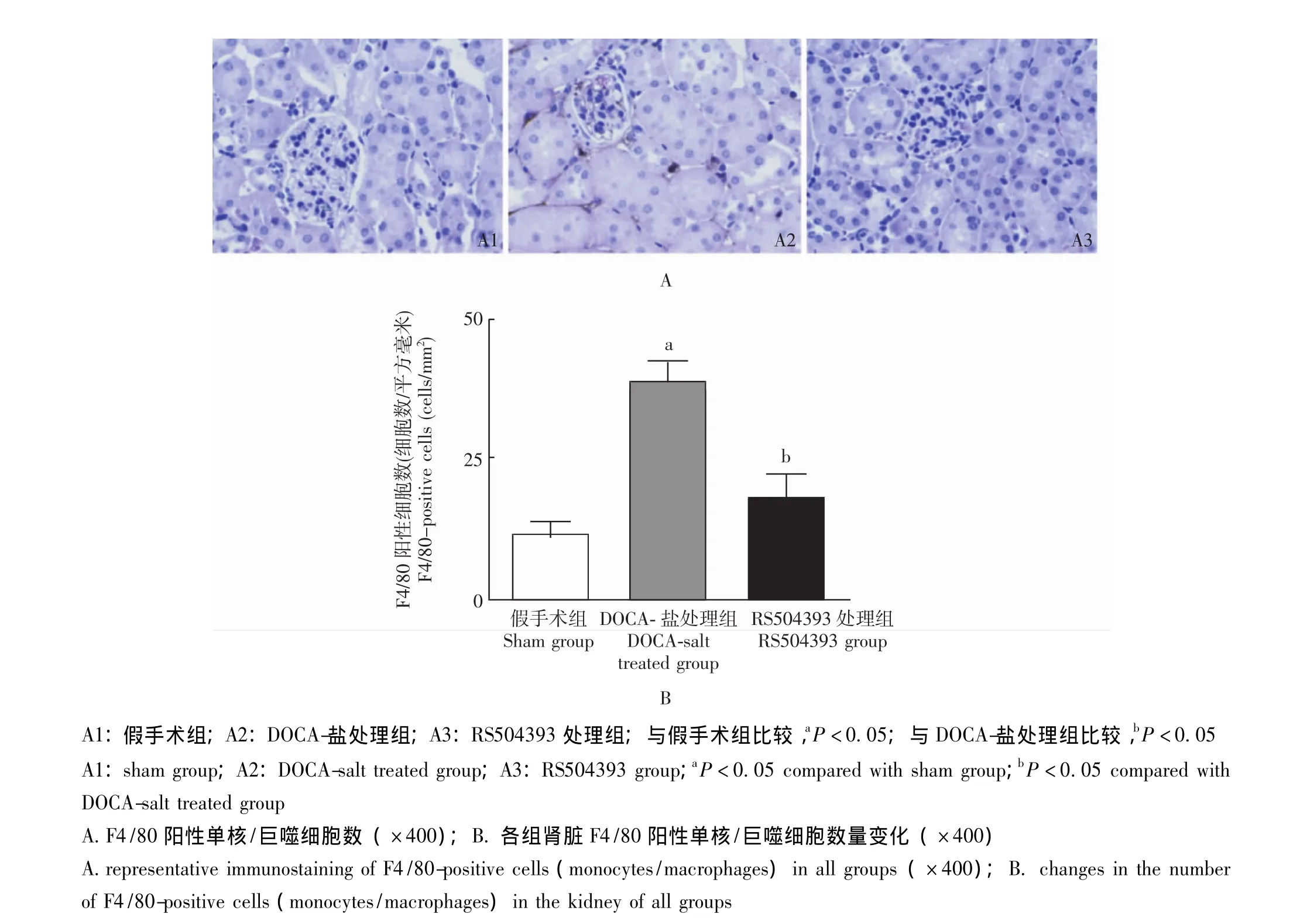
图5 各组小鼠F4/80阳性的单核/巨噬细胞数量比较Fig 5 Changes in renal immunostaining of F4/80-positive cells(monocytes/macrophages)in all groups
高血压介导的肾脏损害往往与单核/巨噬细胞浸润为主的炎症反应和氧化应激反应密切相关[4,14]。浸润入组织的单核/巨噬细胞通过多种因素和机制造成组织损害,其中包括释放各种细胞因子、溶酶体酶、一氧化氮和氧自由基等[15]。来自于单核/巨噬细胞和其他组织细胞的氧自由基可氧化各种细胞成分,如:DNA和细胞膜,从而直接造成组织器官损害。在实验中,目前通常测定8-异构前列腺素以判断氧化应激反应程度,从而间接反映体内氧自由基水平。本研究结果显示,通过皮下包埋DOCA和饮用盐水诱发的盐敏感性高血压造成肾脏损害,在此过程中同时伴随有尿中8-异构前列腺素排泄的增加。另外,CCR2受体阻断剂介导的肾脏保护作用伴随有单核/巨噬细胞浸润的下降及尿中8-异构前列腺素排泄的下降。因此,推测在盐敏感性高血压过程中,CCR2受体阻断剂介导的肾脏保护作用是通过抑制单核/巨噬细胞功能,而其中机制之一可能是通过抑制单核/巨噬细胞产生自由基,从而达到其肾脏保护作用。
本研究结果显示,CCR2受体诱导的以单核/巨噬细胞浸润为主的炎症反应在盐敏感性高血压造成的肾脏损害中发挥着重要作用。在盐敏感性高血压过程中,CCR2受体阻断剂通过抑制单核/巨噬细胞的浸润及相关活性物质的产生,如:氧自由基的释放,从而达到其肾脏保护作用。因此,临床上通过药理学方法阻断CCR2受体介导的信号转导通路可能成为防治盐敏感性高血压造成肾脏损害的方法之一,从而减少高血压,尤其是盐敏感性高血压诱发的终末期肾脏病的发生。
[1]Morimoto A,Uzu T,Fujii T,et al.Sodium sensitivity and cardiovascular events in patients with essential hypertension[J].Lancet,1997,350(9093):1734-1737.
[2]Jamerson KA,Townsend RR.The attributable burden of hypertension:focus on CKD [J].Adv Chronic Kidney Dis,2011,18(1):6-10.
[3]Rostand SG,Kirk KA,Rutsky EA,et al.Racial differences in incidences of treatment for end stage renal disease [J].N Engl J Med,1982,306(21):1276-1279.
[4]Wang Y,Wang DH.Aggravated renal inflammatory responses in TRPV1 gene knockout mice subjected to DOCA-salt hypertension [J].Am J Physiol,2009,297(6):F1550-F1559.
[5]Niu J,Kolattukudy PE.Role of MCP-1 in cardiovascular disease:molecular mechanisms and clinical implications[J].Clin Sci(Lond),2009,117(3):95-109.
[6]Sayyed SG,Ryu M,Kulkarni OP,et al.An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes[J].Kidney International,2011,80(1):68-78.
[7]Hashimoto J,Ito S.Central pulse pressure and aortic stiffness determine renal hemodynamics:pathophysiological implication for microalbuminuria in hypertension [J].Hypertension,2011,58(5):839-846.
[8]Van Liew JB,Davis FB,Davis PJ,et al.Calorie restriction decreases microalbuminuria associated with aging in barrierraised Fisher 344 rats [J].Am J Physiol,1992,263(3 Pt 2):F554-F561.
[9]Springate JE,Feld LG,Ganten D.Renal function in hypertensive rats transgenic for mouse renin gene[J].Am J Physiol,1994,266(5 Pt 2):F731-F737.
[10]Nagase M,Shibata S,Yoshida S,et al.Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker [J].Hypertension,2006,47(6):1084-1093.
[11]Mundel P,Shankland SJ.Podocyte biology and response to injury [J].J Am Soc Nephrol,2002,13(12):3005-3015.
[12]Rhaleb NE,Pokharel S,Sharma U,et al.Renal protective effects of N-acetyl-Ser-Asp-Lys-Pro in deoxycorticosterone acetate-salt hypertensive mice [J].J Hypertens,2011,29(2):330-338.
[13]Wang Y,Wang DH.Protective effect of TRPV1 against renal fibrosis via inhibition of TGF-β/Smad signaling in DOCA-salt hypertension [J].Mol Med,2011,17(11-12):1204-1212.
[14]Tian N,Moore RS,Phillips WE,et al.NADPH oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension [J].Am J Physiol,2008,295(6):R1858-R1865.
[15]Rees AJ.Monocyte and macrophage biology:an overview[J].Semin Nephrol,2010,30(3):216-233.



