谢 骏,屈 岭,梁晓春
中国医学科学院 北京协和医学院 北京协和医院中医科,北京 100730
自噬已经被证实与肿瘤、神经退行性病变、糖尿病等多种疾病的发生密切相关[1- 2]。研究表明,糖尿病患者的机体可以通过自噬防御途径减轻氧化应激损伤,减缓多种慢性并发症的发生和进展[3]。糖尿病神经病变(diabetic neuropathy,DN)是一种由糖尿病引起的中枢神经、周围神经以及自主神经系统病变的症候群[4],在1型与2型糖尿病患者中均可发生。DN初期典型表现为感觉神经病变,包括对触觉、震动、针刺、冷刺激、热刺激的敏感性降低甚至感觉丧失等阴性症状,同时也包括痛觉过敏的阳性症状。晚期病变损伤到运动神经纤维会导致肌肉无力,甚至瘫痪[5]。由于DN患者神经组织长期处于缺血缺氧、营养物质和神经因子缺乏的状态下,加之持续存在的氧化应激损伤共同构成了自噬功能异常的前提和基础[3]。近年来一系列体内外实验研究均证实了自噬与DN之间有着密切的联系,然而二者之间的具体作用关系仍不明确。本文总结了自噬与DN发病机制的研究进展。
自噬及其发生机制
自噬是一种自我降解的过程,是细胞中受损的细胞器和错误折叠蛋白运输到溶酶体内消化降解的过程。自噬在维持细胞内环境稳态中起到了双面调节的作用:正常水平的自噬可以保护细胞免受环境刺激的影响,但自噬过度和自噬不足却可能导致疾病的发生[6]。一旦自噬功能受到破坏,机体神经系统会发生各种病理学改变,产生包括神经退行病变在内的一系列疾病[2,7]。
根据细胞物质输送至溶酶体的方式不同,自噬可分为巨自噬、分子伴侣介导的自噬(chaperone-mediated autophagy,CMA)和微自噬3类;巨自噬是指胞质内粗面内质网、高尔基体等来源的杯状分隔膜包绕损坏的细胞器和可溶性蛋白质形成自噬体,自噬体通过细胞骨架的微管系统运输至溶酶体后,其外层膜与溶酶体膜融合成自噬溶酶体,经过囊泡酸化后被多种蛋白酶降解的过程[8- 9];CMA仅存在于哺乳动物中,胞质中的热休克同源蛋白(heat shock cognate protein,Hsc 70)携带的可调控的分子伴侣会特异性识别包含赖氨酸-苯丙氨酸-谷氨酸-精氨酸-谷氨酰胺的五肽基序蛋白,与其形成复合物后再与溶酶体膜受体蛋白2A(lysosomal associated membrane protein 2A,LAMP- 2A)对接,使底物构象改变成多聚体后转运至溶酶体,进一步激活自噬通路受体,在溶酶体内热休克蛋白90(heat shock protein 90,HSP90)的介导下,底物进入溶酶体腔被蛋白酶分解[8,10- 11];微自噬是指溶酶体在细胞内容物旁边形成内陷,直接吞噬细胞器或其他胞质内容物,并将其降解再利用的过程[12]。
DN中的自噬调节失衡现象
由于细胞自噬既是一种广泛存在的正常生理过程,又是细胞对不良环境的一种防御机制[13],自噬在DN中到底扮演着怎样的角色仍不明确,可能取决于细胞当时所处环境下的病理状态。研究发现,糖尿病大鼠的坐骨神经轴突横断面[14]和背根神经节的感觉神经元[15],代谢综合征的大鼠坐骨神经[16]及外周神经的交感神经轴突中均可发生自噬[17]。同样,在胰岛素导致的低血糖模型大鼠中,早期病变和晚期的再生轴突中均出现了明显的自噬相关结构[18]。因此,自噬是参与血糖改变引起的神经病变的发生发展过程中一个重要环节,糖尿病引起的神经病变多伴随一系列的自噬调节失衡现象。
DN引起自噬减少目前大多研究者均认为糖尿病或高糖环境会降低组织或细胞的自噬水平,从而引发神经组织产生各种病理改变。通过诱导激活自噬,可以减缓这一系列病理生理学改变的过程,从而达到改善DN的效果。
体外实验表明,高糖环境模拟的糖尿病模型中神经细胞的自噬水平受到抑制:Yerra等[19- 20]研究认为,自噬可以通过减少神经细胞内受损的细胞器与蛋白的堆积发挥神经保护作用,高糖培养的neuro2a(N2A)细胞相比正常培养的细胞中自噬体形成减少,并伴有Beclin1、LC3-Ⅱ蛋白表达的减弱。Qu等[21- 22]研究发现,高糖环境下培养24 h后,永生化大鼠雪旺细胞(Schwann cells,SCs)系RSC96细胞和大鼠原代SCs中自噬体均较正常对照组明显减少,增殖活性受到明显抑制,Beclin- 1和LC3蛋白的表达量显着降低。
动物实验中也有类似报道:Yang等[23]研究发现,在24周链脲佐菌素(streptozotocin,STZ)诱导的DN大鼠模型中,小脑中浦肯野细胞退化,伴有轴突末端渐进性膨胀,自噬体形成减少,溶酶体相关膜蛋白2(lysosome associated membrane protein 2,LAMP2)表达量与LC3Ⅱ/LC3Ⅰ比值降低,自噬的特异性降解P62蛋白聚集增多。同样,在STZ诱导的DN大鼠的坐骨神经中Beclin- 1的表达也明显减少[19- 20]。吴琳[24]认为糖尿病与中枢神经系统疾病密切相关,STZ诱导的糖尿病抑郁症大鼠的海马中枢神经元损伤表现为氧化应激水平升高,神经元凋亡,内质网应激水平升高,自噬水平降低,大鼠海马组织中自噬溶酶体数量明显减少,Beclin- 1蛋白表达减弱,P62蛋白表达增强。
同样,在临床研究中也有类似发现:Mohseni等[25]对伴有神经病变的糖耐量减低和2型糖尿病患者进行了长达11年的纵向研究,他们评估了患者腓肠神经纤维病变与自噬相关结构(溶酶体、自噬前体膜、自噬体、自溶酶体样结构及不同降解阶段的细胞器等)之间的病理学关系,结果发现,在基线水平状态下,2型糖尿病患者较正常糖耐量和糖耐量减低患者腓肠神经振幅明显降低,2型糖尿病和糖耐量减低患者的有髓鞘轴突直径更短;11年后发展成2型糖尿病的正常糖耐量人群和糖耐量减低人群与11年后仍保持正常糖耐量的人群相比 g-ratio(轴突直径与髓鞘外径的比值)更高。在随访过程中,2型糖尿病患者的神经传导功能、神经振幅、髓鞘纤维密度、无髓轴突直径、有髓神经轴突中自噬相关结构与基线水平相比均明显降低。这些研究结果说明,2型糖尿病伴发神经病变的病理机制与糖尿病患者腓肠神经发生自噬水平降低相关。
DN引起自噬增多虽然大部分学者支持糖尿病会抑制神经组织或神经细胞的自噬,然而也有部分学者提出了不同的观点。刘晓星[26]研究发现,高糖环境培养72 h后,RSC96细胞可以在引起细胞凋亡损伤的同时,上调细胞中的自噬相关蛋白LC3、Beclin- 1、Atg5、Atg7和FoxO3的表达,激活自噬的腺苷酸活化蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)途径的上游激酶转化生长因子-β激活激酶1(transforming growth factor-β-activated kinase1,TAK1),该结果提示高糖环境下RSC96细胞发生凋亡并伴随自噬的过度激活。上述不同的研究结果可能与高糖作用时间不同相关,高浓度葡萄糖短时间的作用环境下可能引起自噬减少,而长时间作用下可能会引起自噬增加并产生凋亡,但具体的作用浓度与时间的关系有待进一步探究。
Towns等[27]认为自噬是机体对DN引起的恶性环境产生的重要应激反应,其在体内实验发现STZ诱导的DN大鼠模型背根神经节神经元表现出线粒体功能受损,凋亡增加,自噬增强,并且神经元胞体中与线粒体共定位的自噬体增多;其在体外实验中则发现伴有神经病变的2型糖尿病患者血清可以增强人神经母细胞瘤SH-SY5Y细胞中LC3-Ⅱ和Beclin- 1的活性,促进细胞中自噬体的聚集并伴有增大的自噬空泡。该过程可能是通过IgM或IgG自身免疫球蛋白刺激自噬增加,减少线粒体合成,诱导神经元细胞发生自身免疫介导的细胞应激和细胞凋亡来实现的。
Towns等[28]还进一步研究发现,伴有神经病变的2型糖尿病患者的血清中浓缩的免疫球蛋白片段可以诱导SH-SY5Y细胞产生与Fas相关死亡结构域蛋白结构(Fas associated protein with death domain,FADD)共定位的自噬结构;在SH-SY5Y细胞中加入Fas激动剂同样也可以激活自噬的产生;但将血清经可溶性Fas受体嵌合体预处理后则不能产生激活自噬的作用。该结果显示,伴有神经病变的2型糖尿病患者血清中的自身免疫球蛋白可以激活Fas受体复合物并且体外诱导SH-SY5Y细胞产生自噬,表明自身抗体激动剂可以通过刺激Fas串联系统为免疫系统与自噬的激活之间提供了桥梁,同时证实了死亡受体信号通路也参与其中。
Towns等[29]认为Fas自身抗体可以激活自噬,对于非凋亡性的细胞死亡具有细胞保护作用,是一种早期细胞保护反应。因此,自噬的激活可能是DN的病理机制之一。然而,由于其实验是将伴有神经病变的2型糖尿病患者的血清提取物加入SH-SY5Y细胞中进行体外培养,是将细胞加入外来的异种蛋白引起自身免疫反应而诱发的自噬,因此这一过程与机体自身糖尿病高糖环境所产生的病理变化所导致的自噬存在差异。
不同类型糖尿病对自噬的影响不同Osman等[7]用电镜观察了正常人和1型、2型糖尿病患者周围神经病变行腕管松解术术后骨间后神经中的自噬相关结构(吞噬泡、自噬体、自噬溶酶体、溶酶体的电子密度等)的超微形态,结果发现3组人群前臂的骨间后神经中均可见到自噬相关的超微结构,且1型糖尿病患者骨间后神经组织中自噬相关的超微结构数量明显多于2型糖尿病患者,但二者与正常对照组都没有明显差异,推测可能与患者数量不足有关。他们还对3组患者骨间后神经的束肌面积、有髓鞘神经纤维直径与密度、无髓鞘神经纤维直径与密度、有髓鞘纤维的g-ratio进行定性和定量评价,结果发现正常对照组中仅有少量有髓鞘轴突退化和再生,糖尿病组的有髓鞘轴突大量消失或缩小,其中1型糖尿病患者比2型糖尿病患者表现更加明显。这一研究直观表明了DN可能与自噬相关,且1型与2型糖尿病中自噬的调节方式可能不同。但自噬发生是否与糖尿病的类型直接相关仍需进一步探究,不能排除自噬与不同类型糖尿病时机体的血糖状况、胰岛素水平以及病程进展过程中所造成的内环境不同有关。
DN影响自噬调节的机制与治疗
目前自噬参与DN发生发展过程的机制仍不明确,有学者认为,高糖环境会通过影响与自噬相关的代谢通路引起自噬的调节功能失常,并同时影响线粒体合成与分解、神经营养因子的释放、SCs的生长、髓鞘与神经元轴突的修复与再生等一系列病理生理过程影响DN。
自噬与腺苷酸活化蛋白激酶/雷帕霉素靶蛋白通路自噬受到多条信号通路调控,腺苷酸活化蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)/雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路是其中重要一条[30]。高糖环境会通过损伤AMPK/mTOR信号通路抑制自噬,导致线粒体合成受阻,引起感觉神经元功能障碍。活化的AMPK/mTOR通路可以促进自噬形成,通过清除受损的线粒体增强线粒体功能,激活细胞内分解途径产生ATP,减轻DN的炎症反应。Yerra等[19]研究发现,AMPK激活剂A769662可以通过促进LC3B-Ⅰ分裂为LC3B-Ⅱ,增强Beclin- 1的表达,增加自噬体数量,加速机体对损坏或老化线粒体的清除,同时激活过氧化酶增殖因子活化受体γ共同激活剂1 α(peroxisome proliferator-activated receptor gamma,coactivator 1 alpha,PGC- 1 α)介导的线粒体生物合成途径,提高线粒体相关功能;同时A769662还可以抑制核因子κB(nuclear factor kappa B,NF-κB)介导的神经炎症反应。此外,Yerra等[20]发现异甘草素可以通过活化沉默信息调节因子2相关酶1(silent mating type information regulation 2 homolog 1,SIRT1)激活AMPK介导的自噬反应,抑制高糖环境中N2A细胞mTOR的磷酸化,上调自噬相关蛋白Beclin- 1、Atg7和LC3-Ⅱ的表达诱导自噬产生;在体内试验中,异甘草素可以增加STZ模型大鼠外周坐骨神经中烟酰胺腺嘌呤二核苷酸氧化态(nicotinamide adenine dinucleotide+,NAD+)/烟酰胺腺嘌呤二核苷酸还原态(reduced form of nicotinamide-adenine dinucleotide,NADH)来调节SIRT1的表达,激活SIRT1/AMPK介导的自噬途径缓解STZ大鼠的高糖毒性,改善神经病变的功能学和行为学指征。
自噬与线粒体调控途径线粒体自噬是一种细胞内部对功能失调线粒体的自我吞噬,可以帮助解除细胞氧化应激状态并维持线粒体正常的生物合成,同时阻止线粒体毒性物质向胞内释放,维持细胞的正常存活[31- 32]。线粒体自噬可以对多种神经退行性病变起到保护作用,因此调节线粒体自噬功能也被认为是一种治疗DN的新型治疗方向[33]。
Yerra等[34]认为慢性高糖状态下细胞自噬通路受损会导致神经元细胞内突变蛋白的异常堆积以及外周神经系统线粒体功能紊乱。增强神经元细胞自噬活性可以帮助维持线粒体正常形态与功能,阻止活性氧产生与受损蛋白聚集,缓解细胞的生物学功能障碍,减少细胞坏死与凋亡产生,同时可以阻止高糖环境培养的神经元细胞和SCs中毒性物质聚集。PGC- 1 α介导的线粒体能量代谢通路与自噬密切相关,异甘草素可以通过激活SIRT1诱导自噬,同时进一步活化PGC- 1α介导的线粒体合成路径,调控叉头转录因子O3a(forkhead box O3a,FOXO3a)介导的抗应激性,减轻高糖环境中N2A细胞的活性氧产生与线粒体膜去极化,增强线粒体合成,改善线粒体功能[20]。
Srinivasan等[35]在动物实验中发现,慢性高糖环境下大鼠背根神经节神经元细胞中堆积了大量去极化的线粒体并引发细胞凋亡。Towns等[27]研究结果显示,STZ诱导的DN模型大鼠背根神经节神经元细胞胞体中自噬标记抗体(anti-LC3)与线粒体标记抗体(anti-AMA)共定位的荧光强度较正常大鼠明显增加,提示糖尿病大鼠中自噬过程伴随着线粒体能量合成。Yang等[23]研究证实,在24周STZ诱导DN模型大鼠中,过度激活的亮氨酸富集重复激酶2(leucine-rich repeat kinase 2,LRRK2)会在抑制小脑中浦肯野细胞自噬体形成的同时,伴有LRRK2介导的tau蛋白的高度磷酸化和线粒体动力学蛋白(mitochondrial dynamin-like protein,mito-DLP1)高表达,减少线粒体蛋白降解,导致线粒体碎裂,破坏线粒体功能。该实验证实了STZ诱导的DN模型大鼠表现的运动缺陷可能与LRRK2诱导的线粒体功能障碍和小脑浦肯野神经元丢失有关。
因此,通过针对线粒体自噬的分子靶标药理学干预建立线粒体蛋白稳态,改善DN中的代谢失调和线粒体紊乱现象,为治疗DN提供了新思路。
自噬与脑源性神经营养因子脑源性神经营养因子(brain-derived neurotrophic factor,BNDF)参与调节多种神经功能,包括影响神经元、轴突、树突分支[36- 37]以及突触的形成、成熟与稳定[38- 39]。除了已确立的神经营养作用外,BDNF还具有其他神经保护作用,包括抗凋亡、抗氧化和调节自噬[40]。
Bak等[41]研究发现,可以内源性合成ω3多不饱和脂肪酸(ω3 polyunsaturated fatty acids,ω3-PUFA)的fat- 1转基因小鼠较野生型STZ模型小鼠不易发展成高血糖、运动缺陷,并且表现出浦肯野细胞保护作用。因为ω3-PUFA不仅可以上调STZ模型小鼠小脑中BDNF的表达,还可以进一步诱导LC3Ⅱ、Beclin- 1蛋白表达,降低P62水平,增强自噬体成熟相关蛋白Rab7、Cathepsin D和ATP6E的表达。该研究结果提示ω3-PUFA可以增强神经营养因子表达,促进自噬体的成熟,增强小脑浦肯野细胞自噬流活性,保护了浦肯野细胞的存活。因此,ω3-PUFA可以作为治疗伴有运动缺陷的DN新方法。吴琳[24]认为硫化氢可以通过BDNF/TrkB通路促进糖尿病模型大鼠海马神经元的自噬作用,加入BDNF/TrkB通路抑制剂K252a与NaHS可以逆转硫化氢的自噬促进作用,说明BNDF与自噬之间存在着互相介导的关系。
然而在正常生理环境中,有学者认为神经营养因子可以激活PI3K/Akt/mTOR信号级联,从而抑制自噬[42]。Smith等[43]研究发现,BDNF可以通过抑制自噬促进海马神经元细胞存活,这种保护作用可以被mTOR抑制剂雷帕霉素和白细胞介素- 1β阻断,说明BNDF可以通过激活mTOR抑制自噬。由于目前缺少在糖尿病模型中对神经营养因子与自噬发生关系的相关研究,仅有的少量葡萄糖代谢研究认为,糖尿病会导致大脑中胆固醇合成水平降低,在胆固醇减少的GT1- 7下丘脑细胞中,神经营养因子水平降低,基线自噬水平升高,但随着细胞血糖剥夺加重,自噬合成逐渐受阻[44],提示自噬水平是随血糖波动而发生动态变化。糖尿病疾病状态下神经营养因子与自噬发生机制尚无直接研究报道,我们根据现有研究结果仅可以推测,在生理状态下BDNF可通过诱导PI3K/Akt/mTOR通路抑制自噬的产生,但在DN模型中神经营养因子与自噬通路的具体关系,以及其与血糖变化动态过程中不同时期自噬水平的调控关系仍有待进一步阐明。
自噬与SCs和髓鞘蛋白SCs是周围神经系统的髓鞘形成细胞,SCs的正常发育分化及围绕神经元轴突形成髓鞘是外周神经结构和功能成熟的前提;SCs与神经元生长密切相关,其不仅能够分泌神经营养因子,还参与朗飞氏结的形成,形成跳跃式传导的结构基础[45]。研究表明,高糖环境会引起SCs功能障碍[21- 22,46],并且SCs的病变很可能是DN神经再生障碍的始动因素[47- 48]。因此,SCs与DN引起的髓鞘障碍和髓鞘化密切相关[49]。
Qu等[21- 22]研究发现,在高糖环境下培养24 h后,RSC96细胞和大鼠原代SCs的自噬与增殖活性均受到抑制。加入自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)后,SCs表现出随时间和浓度依赖性增殖抑制。中药复方筋脉通的含药血清和槲皮素均可以改善这种高糖环境下SCs的自噬与增殖受抑制现象,减少细胞损伤。动物实验研究发现,中药复方筋脉通不仅可以改善DN大鼠坐骨神经的病理形态学改变,还可增加坐骨神经中自噬结构数量,上调Beclin- 1蛋白表达。然而,刘晓星[26]研究发现,高糖环境培养72 h后,RSC96细胞可以在引起细胞凋亡损伤的同时伴有自噬过度激活。使用中药复方糖痛方干预后,RSC96细胞自噬相关蛋白表达下调,表明中药复方糖痛方可能是通过抑制细胞的过度自噬减少SCsⅡ型程序性死亡,从而缓解DN。
Hamilton等[50]研究认为,外周髓鞘蛋白(peripheral myelin protein- 22,PMP22)的异常堆积是DN发生的重要病理过程。PMP22蛋白是髓鞘磷脂的重要组成成分,正常神经元细胞中80%新合成的PMP22蛋白都会自行降解,但DN模型小鼠中由于自噬和溶酶体系统的缺陷会导致PMP22不能充分降解而在机体产生异常的堆积。因此,增强自噬可以作为治疗异常蛋白堆积导致DN的新思路。
总结与展望
综上,现有研究中大多学者的观点认为高糖环境可以抑制神经元及神经胶质细胞或机体神经组织中自噬的产生,其作用机制与高糖环境下代谢途径异常、线粒体功能紊乱、SCs增殖受阻、神经营养因子分泌不足、髓鞘脱失及修复再生障碍有关。DN高糖环境下机体代谢相关通路损伤抑制自噬,自噬障碍导致的线粒体功能失常进一步引发细胞凋亡;DN高糖环境下SCs自噬减少,细胞增殖活性下降,分泌神经营养因子不足,神经营养不足导致髓鞘脱失;自噬不足引发外周髓鞘蛋白异常堆积,脱失的髓鞘无法修复再生,导致一系列DN的病理与临床表现(图1)。通过诱导自噬可以缓解DN的症状,控制疾病的进展。
但是也有少部分研究发现高糖环境会促进神经组织和细胞自噬活性增加。由于研究者们的实验方法以及作用时间具有差异性,所以得出了不同的结论。刘晓星[26]的实验条件为高糖作用晚期,RSC96细胞的自噬被过度激活,提示自噬程度可能与高糖的作用时间有关:在高糖干预早期,细胞正常自噬活动被抑制,随着高糖干预时间延长,大量代谢产物逐渐堆积,促使自噬反而被过度激活,诱导细胞发生Ⅱ型程序性死亡。Towns等[27]实验方法是向细胞中加入外源性DN患者的血清提取物,诱发细胞自身免疫反应而产生自噬,该实验过程是否符合机体自身高糖环境所产生的病理变化诱导的自噬需进一步探究。
同时尚有学者认为DN的自噬水平表现出双面性,可能与不同的高糖环境、干预方式以及不同类型的糖尿病等条件有关。不过此部分实验仅限于临床少样本量的观察,缺少统计学意义。虽然目前学术界对自噬与DN的关系存在不同的看法,但大部分学者基于体内外实验验证的结果认同DN时自噬减少的观点。对于持不同观点的学者们因实验方法的不同、模型的差异、观测时间的长短,以及样本量的限制,得出的结论是否绝对与前者相反还有待进一步证实。但也不能排除随着糖尿病发展的不同阶段和机体血糖水平的波动与变化,体内自噬发生水平也有可能随之发生动态改变。因此,究竟自噬是一种潜在的细胞存活机制还是导致细胞死亡或疾病发生的病理性机制,或者是同时兼具两种作用,目前尚无定论。
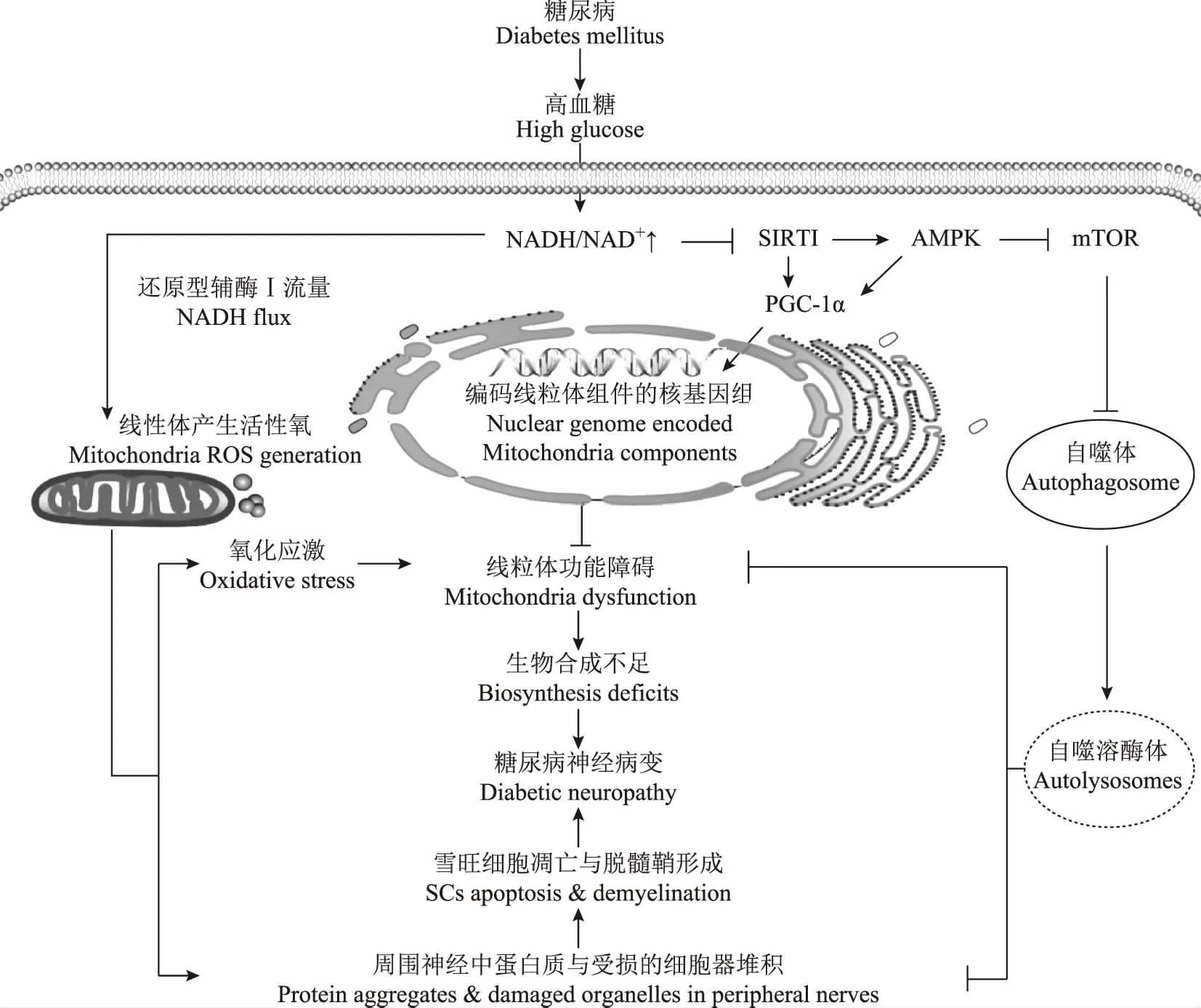
NADH/NAD+:烟酰胺腺嘌呤二核苷酸还原态(还原型辅酶Ⅰ)/烟酰胺腺嘌呤二核苷酸氧化态;SIRT1:沉默信息调节因子2相关酶1;PGC- 1 α:过氧化酶增殖因子活化受体γ共同激活剂1 α;AMPK:腺苷酸活化蛋白激酶;mTOR:雷帕霉素靶蛋白
NADH/NAD+:reduced form of nicotinamide-adenine dinucleotide/nicotinamide adenine dinucleotide+;SIRT1:silent mating type information regulation 2 homolog 1;PGC- 1 α:peroxisome proliferator-activated receptor gamma coactivator 1 alpha;AMPK:adenosine monophosphate-activated protein kinase;mTOR:mammalian target of rapamycin
图1糖尿病神经病变中自噬可能的调控机制
Fig1The potential regulatory mechanisms of autophagy in diabetic neuropathy
尽管自噬对DN的作用仍存在争议,但自噬在DN的病理生理过程中发挥的潜在作用绝对不容忽视。能否通过研制靶向作用于自噬调节机制的新型药物来治疗DN将会为现有的治疗模式提供新的思路,为DN患者提供新的治疗方案。当前大多数西药治疗DN主要通过纠正代谢紊乱等原发病、促进神经修复、抗氧化应激、改善微循环、醛糖还原酶抑制剂等治疗方法来控制DN的进展[51],直接作用于自噬途径治疗DN的药物还鲜有报道。少数学者发现自噬相关通路的激活剂具有一定改善DN的疗效,如AMPK激活剂可以通过激活AMPK/mTOR信号通路促进自噬和线粒体合成,抑制NF-κB介导的神经炎症反应[30]。此外,通过调控自噬与神经营养因子分泌的药物也可以促进神经修复,发挥一定的神经保护作用,如ω3多不饱和脂肪酸可以促进自噬上调神经营养因子表达改善神经修复功能[41],硫化氢可以通过促进神经营养因子表达诱导自噬形成保护神经元功能[24]。但是文献报道数量有限,研究内容形式也需要进一步扩充。在中医药领域,由于中药在治疗疾病中具有多靶点多方向的网络式调节特点,其在自噬与DN研究领域中的疗效引起了学者们更为广泛的关注。现有研究发现,异甘草素、槲皮素、筋脉通、糖痛方等中药单体或中药复方在一系列体内外实验中均可以通过调节机体自噬功能改善DN。但是,现有文献得出的结论尚不完全统一,同时也缺乏深入的分子生物学及动物实验对机制进行详细阐述,更缺少大规模、规范化的高质量临床随机对照双盲研究。
因此,进一步开展深入的临床与基础研究,探索自噬与DN之间的作用机制,对于寻找DN新药物和新靶点具有深远的意义。大规模多中心的临床随机对照试验观察DN患者神经组织中自噬水平的变化,探讨不同类型糖尿病患者神经病变与自噬的关系,及时监测患者在疾病不同发展阶段自噬水平的动态改变是临床研究中亟待解决的问题。目前临床已有报道利用角膜激光共聚焦显微镜检查技术(invivocorneal confocal microscopy,IVCCM)无创检测DN患者角膜神经退化与神经元纤维的病理结构改变[52- 53],这无疑为DN的早期筛查和诊断,以及大规模临床试验研究的开展提供了便利。基于IVCCM技术的DN患者自噬水平变化研究目前未见报道,可以作为开展DN临床研究的新方向。同样,深层次的基因分子生物学实验探究DN患者及动物模型中相应神经组织中自噬相关结构的变化也迫在眉睫。从基因组学、转录组学、蛋白质组学、代谢组学等不同层面探讨自噬与DN的病理生理学改变机制,研究自噬对DN机体代谢途径、线粒体调节、神经营养因子分泌、SCs生长、髓鞘脱失与修复再生等方面的影响,同时检测造模不同时间段中,随DN进展的不同时期血糖浓度不断变化时,机体自噬水平发生的动态改变,也是研究自噬与DN之间相互作用关系的重要环节和未来探索自噬调控DN发生发展机制的重点方向。



