,,5
(1 西安交通大学附属儿童医院(西安市儿童医院)心内科,陕西 西安 710003; 2 空军军医大学第一附属医院妇产科;3 西安交通大学附属儿童医院(西安市儿童医院)超声科; 4 陕西省儿科疾病研究所; 5 西安市儿童健康与疾病重点实验室)
糖原贮积病Ⅱ型(GSDⅡ)又称庞贝病,发病率约为1/40 000[1],是一种罕见的常染色体隐性遗传病。其发病机制为位于17q25.3染色体上编码酸性α-葡糖苷酶(GAA)的基因发生突变,使溶酶体内GAA缺乏,致糖原不能转化为葡萄糖,从而在骨骼肌、心肌和平滑肌等组织细胞内大量沉积[2]。本研究对2016年12月—2017年5月于西安市儿童医院心内科确诊的4例伴有肥厚型心肌病的GSDⅡ病儿的临床特点、GAA活性以及GAA基因进行分析,总结临床表现、酶学特点和基因变异的相互关系,并对下一胎进行遗传咨询和产前诊断。现将结果报告如下。
1 对象与方法
1.1 研究对象
2016年12月—2017年5月西安市儿童医院心内科确诊的GSDⅡ住院病儿4例,男3例,女1例,发病年龄为2~8月龄,确诊年龄为2~9月。4例病儿均因呼吸道感染就诊,有明显的运动发育迟缓或伴语言发育落后,主要为四肢无力、肌张力低下,累及四肢、躯干肌,面肌未受累。4例呼吸肌无力,均有心肌受累;舌体肥大2例;4例伴有肝大。4例肌酸激酶(CK)、谷丙转氨酶(ALT)、谷草转氨酶(AST)均升高,达正常值3~10倍。4例进行了心电图检查,提示PR间期80~90 ms,双心室肥厚。1例肌电图检查示肌源性损害。4例病儿心脏B超均示左心室增大,左心室壁弥散性增厚,左心室后壁内膜增厚,提示肥厚型心肌病。病儿分别来自4个不同家庭,父母均非近亲婚配,无遗传病家族史,因呼吸道感染、心脏超声发现心肌肥厚就诊。临床资料包括病史、心电图、超声心动图、心脏核磁共振、心肌酶、肝肾功及GAA酶活性检查和基因检测。本研究经西安市儿童医院伦理委员会批准同意,病儿家长均签署了知情同意书。
1.2 研究方法
1.2.1GAA酶活性测定 外周血白细胞用生理盐水重悬,取20 μL白细胞悬液(约2 μg蛋白)加入80 μL反应体系(1 mmol/L 4-甲基伞形酮-α-D-葡萄糖苷,pH 4.3的50 mmol/L醋酸钠缓冲液,1 g/L的Triton X-100,10 μmol/L阿卡波糖)中,37 ℃水浴反应2 h。加入pH 10.7的0.1 mol/L甘氨酸-氢氧化钠缓冲液150 μL终止反应。检测反应体系荧光(BioTek SynergyH4多功能酶标仪)激发波长360 nm,发射波长450 nm。空白对照为相同条件,反应体系中不加蛋白。配置不同浓度的4-甲基伞形酮,绘制标准曲线,按照标准曲线计算出产物浓度。蛋白浓度用Bradford法测定。
1.2.2基因组DNA的提取和GAA基因突变分析基因组DNA的提取:采集病儿及其父母外周血各2 mL,用血液基因组DNA提取试剂盒(天根生化科技(北京)有限公司)提取基因组DNA,放至-80 ℃低温冰箱保存备用。高通量测序(病儿2):将先证者的基因组DNA送至北京信诺百世医学检验所进行心血管系统疾病相关基因panel的靶向捕获二代测序,对二代测序所分析出的候选基因突变位点在家系内进行Sanger测序验证。Sanger测序(病儿1、3、4):依据NCBI GAA基因 (NM_000152.3)序列,采用Primer 5.0软件设计引物,扩增GAA基因的20个外显子序列。引物由上海生工生物工程有限公司合成。PCR反应条件为:95 ℃ 5 min, 95 ℃ 30 s,55 ℃ 30 s, 72 ℃ 30 s,共进行30个循环,最后72 ℃延伸7 min。PCR扩增产物用20 g/L琼脂糖凝胶电泳检测,之后采用ABI 3500dx仪器进行双向测序。产前基因筛查:针对家庭内有明确基因诊断的病儿母亲在孕7~12周采集绒毛膜,或者于孕16周后行羊膜穿刺,抽取羊水,提取基因组DNA。按照上述Sanger测序的方法,对相应的突变位点所在的外显子进行PCR扩增及Sanger测序,并对测序结果进行分析。
2 结 果
2.1 GAA酶活性
病儿1、2和3 GAA的活性分别为2.5、2.1和1.9 nmol/(g·min),病儿4未行GAA活性测定。见表1。
2.2 GAA基因突变分析
4例病儿均为复合杂合突变,分别来自于病儿父亲和母亲(见表1)。在检测到的8个突变中有5个错义突变,3个插入突变,根据生物信息分析软件SIFT、PolyPhen2及Mutation_Taster致病性分析以及美国ACMG指南[3]预测,其中7个突变为致病性突变,病儿1携带的p.L701P突变预测为可疑致病,结合病儿GAA活性的检测结果,确定为致病性的突变。而在已经检测出的突变位点中,p.L701P、p.W279C、p.N535Qfs*3突变位点在HGMD数据库及相关文献中均未见报道。4例病儿均携带复合杂合突变,符合常染色体隐性遗传,突变测序峰图见图1。
2.3 治疗、随访以及产前GAA基因突变检测
1例病儿接受Myozyme GAA酶替代治疗1个月,其余3 例病儿均未接受酶替代治疗。4例病儿确诊后2~6个月内均死亡,死亡原因为肺部感染、肺不张合,导致呼吸循环衰竭。2例病儿母亲孕下一胎,于妊娠期行羊水穿刺胎儿GAA基因检测,病儿3母亲孕13周绒毛膜致病基因检测确诊胎儿携带GAA c.837G>C和c.1598-1599insC复合杂合突变,终止妊娠。病儿1母亲孕18周羊水穿刺GAA检测确定胎儿携带一个杂合突变,遗传咨询结果考虑胎儿不会发病,现已经足月娩出,出生体质量约3.4 kg,健康。
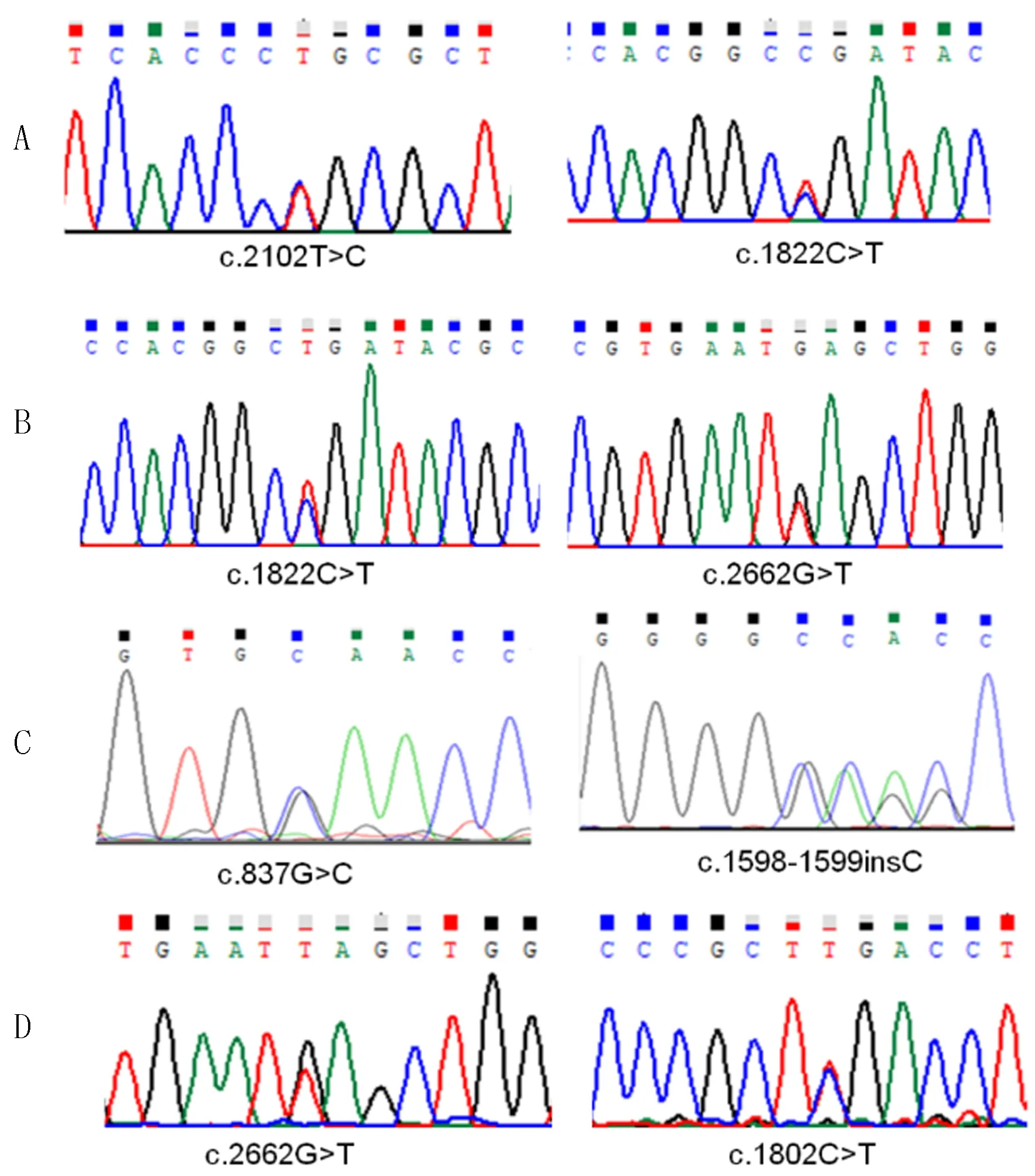
A、B、C、D分别为病儿1、2、3、4。
图14例病儿GAA突变位点测序图
3 讨 论
GSDⅡ分为早发型 (婴儿型)及晚发型 (青年型及成人型)。晚发型GSDⅡ多青少年起病,表现为肌力减退和呼吸肌受累等症状,罕见心肌受累[4]。早发型婴儿期起病,症状严重,心肌受累的超声心动图表现为以左心室心肌肥厚为主的双心室心肌向心性肥厚,多在1岁以内死于心力衰竭。由于婴儿型早期临床表现非特异、进展迅速、诊断治疗时间窗短[5],早期快速做出正确诊断对病儿的治疗和预后有重大意义。本组报道的4例病儿发病年龄均在1岁前,伴有肥厚型心肌病和骨骼肌受累,故为早发型GSDⅡ。
GAA基因突变影响GAA的合成、磷酸化修饰、转运以及分泌,突变性质决定残余GAA活性,GAA活性越低,发病年龄越早,临床表现越严重[6]。婴儿型病儿皮肤成纤维细胞的GAA活性常低于正常对照的1%,而晚发型残余酶的活性可达1%~40%[7]。国内有报道幼年起病的晚发型GSDⅡ型病儿,但无心肌肥厚表现[8]。而本组4例病儿均有明显的心肌肥厚,心功能有不同程度的降低,血肌酸激酶升高较显着,提示该4例病儿属于典型的婴儿型GSDⅡ。
GAA基因的突变型与临床表型相关,在该组病儿1中发现的GAA基因c.1822C>T,p.R608X杂合突变为HGMD报道的已知致病性突变[9-10];而c.2102T>C,p.L701P杂合突变既往未见报道,生物信息分析软件SIFT、PolyPhen2及Mutation_Taster均预测该突变可能有害,病儿1临床发病,证实了p.L701P为致病突变。病儿2携带的GAA基因c.1822C>T,p.R608X杂合变异,为HGMD报道的与GSDⅡ相关的已知变异,人群样本数据筛查在个例病人中检出上述变异[11-14],该变异为无义变异,造成终止密码提前出现,提示该变异可能对蛋白多肽链的正常合成有重要影响,该变异在外显子组整合数据库(ExAC)中的频率为0.00002479,定义为致病性变异。KOBAYASHI等[15]通过将重组GAA (rGAA)对病人进行体内置换治疗,发现可以减轻早期病人的症状。病儿2携带的另外一个GAA基因c.2662G>T,p.E888X杂合变异为HGMD报道的与GSD相关的已知变异[15-18]。p.E888X在ExAC中的频率为0.00002,为致病性变异。本组病儿中有2例携带p.E888X,进一步证实该突变为热点突变。病儿3 GAA外显子4和11分别检测到一个杂合突变位点c.837G>C,p.Trp279Cys和c.1598-1599insC,p.N535Qfs*3,这两个位点在HGMD数据库及相关文献中均未见报道。 c.837G>C突变导致GAA蛋白的第279位氨基酸由色氨酸(Trp)变为半胱氨酸(Cys),通过http://genetics.bwh.harvard.edu/pph2/预测其为可能致病。c.1598-1599-insC为移码突变,导致GAA蛋白翻译到第537位氨基酸时终止,形成一个不成熟的蛋白截短体,影响蛋白正常功能的发挥,通过http://www.mutationtaster.org/预测该位点为极有可能致病。家系验证结果显示,受检者的父亲和母亲分别携带c.1598-1599insC和c.837G>C,符合遗传共分离规律。病儿4样本分析显示GAA基因有2个杂合突变,即c.1802C>T,p.S601L杂合突变以及c.2662G>T,p.E888X杂合突变。这两个位点均为HGMD报道与GSDⅡ相关的疑似致病性突变[19]。
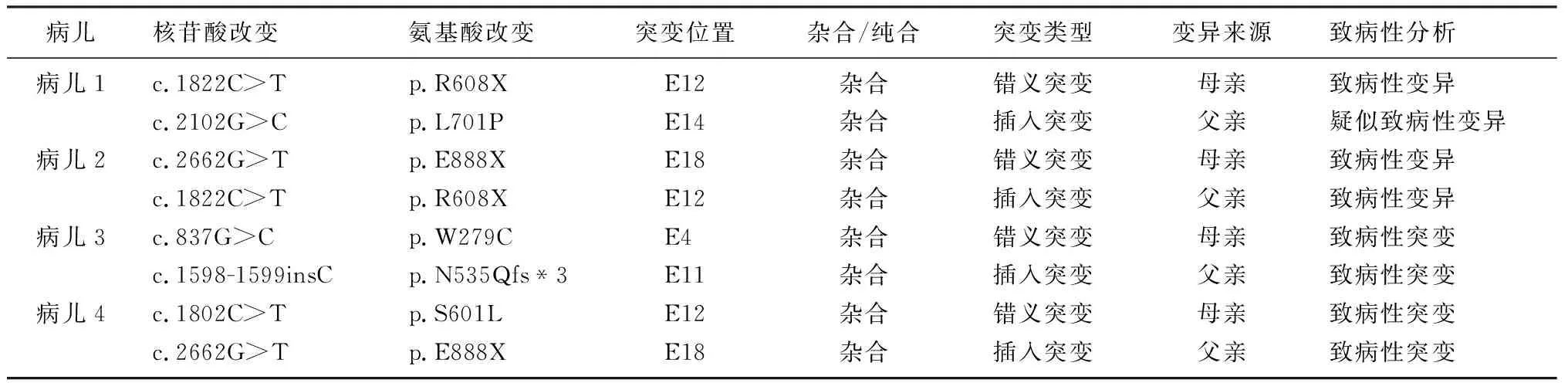
表1 病儿GAA基因突变结果
GSDⅡ为常染色体隐性遗传,病儿分别携带来自父母的致病突变方可发病,绒毛、羊水和胎儿血均可提取胎儿DNA用于产前遗传病诊断。妊娠7~12周可以选择绒毛膜,缩短诊断周期。妊娠14~22周可以选择羊水穿刺,羊水取材安全性高、结果准确。如果妊娠超过22周,可以用胎儿血培养。胎儿血培养成功率高,染色体核型形态好、分裂相多[20]。本组病儿中2例母亲孕下一胎,1例妊娠13周取得绒毛膜进行GAA测序后发现胎儿携带2个致病位点,终止妊娠。 1例在妊娠18周行羊水穿刺,GAA基因检测确定胎儿只携带一个致病基因,继续妊娠,顺利分娩正常新生儿。
综上,婴儿发病GSDⅡ病儿分别携带来自父母的致病突变,病儿症状出现早,未经酶替代治疗死亡率高。GSDⅡ在我国尚未列入产前筛查,对于病儿母亲孕下一胎胎儿进行GAA基因检查,有助于GSDⅡ出生缺陷的预防。
(感谢西安交通大学附属儿童医院(西安市儿童医院)、陕西省儿科疾病研究所杨颖、张李钰对本文基因检测结果分析的帮助)



