胡 坚
自法国学者Hadchouel等[1]首次在儿科报道了幼年类风湿性关节炎合并巨噬细胞活化综合征(MAS)以来,国内外学者对最早由Boone在第一届美国风湿病协会(ARA)会议上所报道的临床资料[2]和不断发现的临床病例进行了大量的临床与试验研究,并将其并入噬血细胞淋巴组织细胞增生症(HLH)的病因分类中[3]。这些发现和临床病理研究,包括对HLH免疫遗传学和免疫病理学的深入研究,逐步加深了临床风湿病学科医生对MAS的认识,早识别、早干预的治疗可明显降低并发此综合征患者的病死率。国内最早的病例报道[4]和随后的集中报道则见于近年[5-7]。作为具有强烈免疫应答复杂背景的MAS多见于近年来被认为固有免疫应答起动为主的Still病,即儿童期全身型起病的幼年特发性关节炎(soJIA)/幼年类风湿性关节炎(JRA)和成人型Still病[6-8],也有系统性红斑狼疮(SLE)和皮肌炎等自身免疫疾病为背景的病例报道[8-11]。由HLH病因分类中可以看出,在众多已经确立的单基因或遗传表型迥异的疾病中,可能存在相关的遗传性或获得性免疫应答缺陷[10]。作为HLH病因分类的特殊原因的自身免疫性疾病,保留了MAS的定义。笔者就作为自身免疫性疾病的MAS的研究谈一点粗浅的认识。
1 MAS与HLH:相似与相同
1.1 临床免疫学 最早的关于吞噬红细胞的网状巨噬细胞在噬血细胞综合征中作用的认识可追溯到20世纪30年代后期,但对衍生于HLH的MAS的病理生理学机制仍然缺乏完整的认识。目前,主要的临床免疫发现是:(1)高水平的T细胞驱使巨噬细胞刺激炎症因子类和细胞因子,包括干扰素(IFN)-γ、单核细胞趋化蛋白(MCP)-1和巨噬细胞集落刺激因子(M-CSF)。(2)表达T细胞核心作用和具有诊断与预后意义的白细胞介素(IL)-2受体(CD25)。(3)部分巨噬细胞驱使的促炎因子,包括IL-6、IL-12、IL-18和肿瘤坏死因子(TNF)-α。(4)在肝组织中发现的与CD68+细胞相关的TNF-α和IL-6的强烈表达[12]。(5)骨髓和淋巴组织中CD163+吞噬血细胞的巨噬细胞显着增殖。(6)并未发现抗原呈递细胞(APCs)在噬血细胞综合征初始阶段的作用[13]。(7)通过CpG DNA刺激Toll样受体(TLR)-9模拟产生MAS的研究提示炎症性疾病合并MAS与慢性固有免疫活化基础有关,而与原发性HLH相关的遗传缺陷无关[14]。在这些发现中,值得关注的是CD163+吞噬血细胞的巨噬细胞显着增殖。通常,CD163是经典意义上的抗炎表型表达,与巨噬细胞分化通路有关。以儿童期Still病/soJIA为例,似乎看上去与其病理作用相悖。(8)在获得性或反应性HLH,NK细胞活性有时会表现出时间性起伏变化(fluctuate over time)。
1.2 免疫遗传学 相比原发性HLH,如家族性穿孔素基因缺陷等,自身免疫病相关的MAS的免疫遗传学研究制约于多因素致病而尚未得到有意义的发现。已确认的遗传性HLH,见表1。代表性的单基因缺陷病虽然部分属于原发性免疫缺陷病(PID),却均可发生HLH。然而,在自身免疫病相关的MAS患者中还未得到相关的遗传学资料。
2 临床特征与诊断
2.1 HLH的基本特征 可以通过2004 HLH的诊断分类和诊断的判定标准获得HLH的基本临床特征[13]。需要指出的是,组织病理发现吞噬血细胞的基本条件是吞噬2系以上血细胞,以区别生理状态下的吞噬清除现象,除常见的骨髓组织以外,脾脏内及血涂片中吞噬血细胞的组织细胞涂片可参见Behrens等[14]的研究报道。同时,组织中未发现吞噬血细胞的现象不能作为否定条件。修正的HLH诊断指南,见表2。
2.2 自身免疫疾病相关的MAS 多种风湿性疾病均有合并MAS的临床报道。主要有皮肌炎、川崎病*、SLE**、成人期起病的Still病、强直性脊柱炎、结节病、炎性肠病、附着点炎关节炎、未分化结缔组织病、多关节型幼年特发性关节炎及儿童期起病的Still病***等(*个数多少代表病例报道集中情况)。在这些疾病中,有的根本就是异质性的。soJIA合并MAS的推荐诊断标准,见表3。当MAS出现时,无论是起病之初,还是在疾病的进程中,原发病的临床表现出现或加强或矛盾的现象时需要临床医生仔细辨别。如Still病合并MAS时,其判定条件的一些项目“不增高就是问题”常是重要的早期发现,包括全血细胞计数的白细胞和血小板计数,以及血沉;中枢神经系统症状的出现常提示其严重性,而血清铁蛋白升高通常又是Still病活动期的特征,因此,还要分析铁蛋白定量水平升高的程度和速率。由于SLE可影响血液系统和神经系统的特殊性,更需要临床医生根据免疫介导的炎症反应指标来进行判断,如血清铁蛋白、血清天冬氨酸转氨酶(AST)和乳酸脱氢酶(LDH)的异常升高等,同时要考虑SLE自身相关的活动性标志[15]。无论哪种风湿性疾病,一旦出现与发热伴随的严重的进行性肝损伤,尤以LDH和AST早期明显升高(可高于正常值数十倍甚至百倍以上)为特征,以及出现凝血机制障碍时,都应考虑MAS可能已发生,而不应简单理解为噬肝病毒感染相关的肝衰竭[12]。

表1 已确认的遗传性HLH

表2 修正的HLH诊断指南
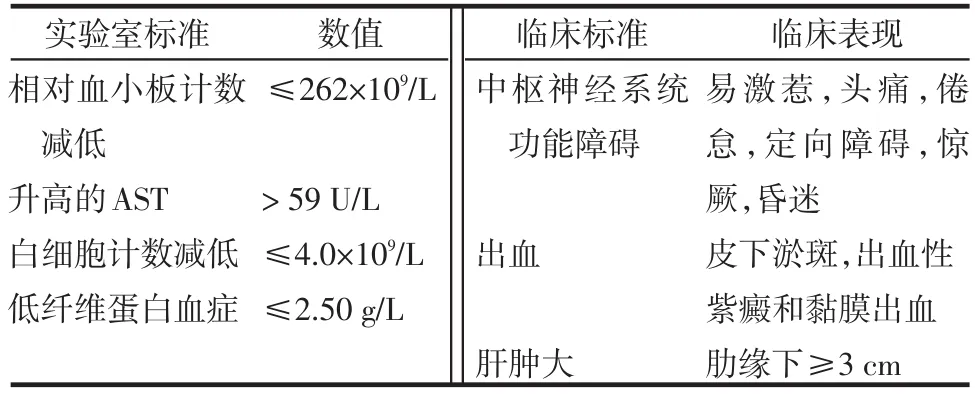
表3 soJIA合并MAS的推荐诊断标准
2.3 相关诱因 无论是原发性HLH还是继发自身免疫病的MAS,感染和药物等相关诱因可能是此综合征重要的患病条件,与HLH有关的促发因素,见表4。甚至是具有诱发自身免疫现象的肺炎支原体感染(肺炎)所表现的严重的、感染相关性HLH在临床也并非少见[16],及时的抗炎治疗能更好地加快临床转归。
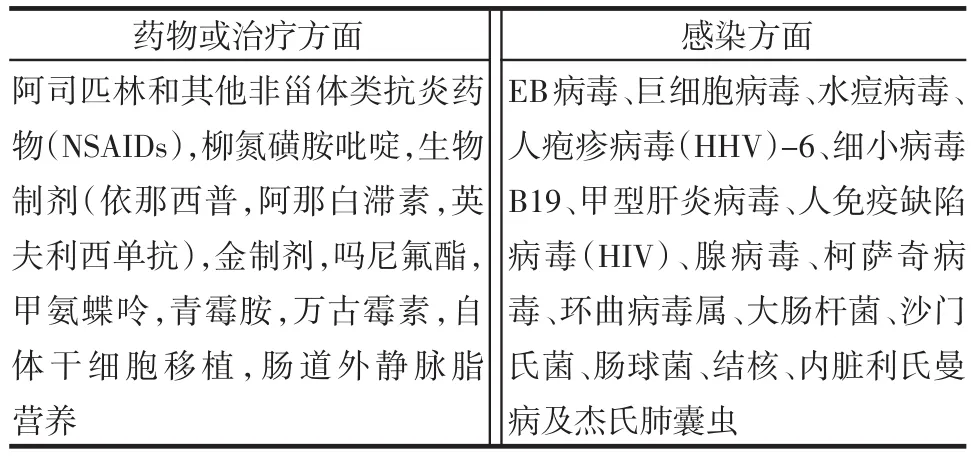
表4 推测与HLH有关的促发因素
2.4 区分疾病背景和临床状态的属性 在确定诊断时,无论是初发病例还是已经获得原发病诊断的病例均需要比较单基因缺陷的原发性HLH诊断标准、继发于自身免疫疾病的MAS,以及感染导致的HLH与系统性炎性反应综合征(SIRS)判定条件间的差异性。特别是感染相关疾病,当患者的临床符合SIRS的判定条件,病程中又出现满足HLH诊断条件的情况时,则应权重考虑基于HLH发病机制的治疗选择。
3 治疗需要权重原发病
尽管临床判定条件已经符合HLH或MAS,但是否采用HLH-04方案[13]仍应权重原发病进行选择。临床基本抗炎药物选择条件是:(1)糖皮质激素(氟美松)抗高细胞因子血症。(2)环孢菌素抑制活化的T细胞并增强糖皮质激素的细胞溶解效应。(3)足叶乙甙(Etoposide)高效诱导单核细胞凋亡。有时调整针对风湿性疾病的免疫抑制剂或大剂量糖皮质激素的冲击以及使用大剂量静脉多价免疫球蛋白即可获得临床缓解,而不需要使用诱导单核细胞凋亡作用较强的足叶乙甙。当出现多器官功能障碍时,配合上述抗炎治疗选择免疫净化(血液净化)或有助于临床转归。IFN-γ阻滞剂或中和抗体,以及TLR和IL-10的调控治疗值得期待[14]。借此次机会提醒临床医生,当原发感染合并HLH时,应认真分析原发感染的证据,既不能随意应用糖皮质激素也不要盲目地提高抗生素等级而放弃基于免疫作用的抗炎药物应用,从而失去最佳治疗时机或导致后遗症。
综上,归类于HLH的自身免疫病相关的MAS正逐渐被更多临床风湿病科医生认识。无论定义为HLH,还是MAS,都与复杂的免疫应答紊乱相关,是一个需要对固有免疫和适应性免疫间相互作用与调节深入研究的临床热点。然而,发生HLH的情况并非局限于风湿性疾病,还涉及包括急救医学在内的多学科相关领域[11],这是一个非常严重的可发生在不同疾病背景下免疫应答紊乱的疾病状态,见表5。延期诊断可致患者短时间内死亡。临床诊断时需要认真考虑此综合征的复杂的异质性疾病背景。早期识别和基于免疫炎症机制的及时治疗可明显改善此综合征患者的预后。

表5 HLH病因
[1]Hadchouel M,Prieur AM,Griscelli C.Acute hemorrhagic,hepatic,and neurologic manifestations in juvenile rheumatoid arthritis:possible relationship to drugs or infection[J].J Pediatr 1985,106(4):561-566.
[2]Ramanan AV,Schneider R.Macrophage activation syndrome-what’s in a name[J]!J Rheumatol,2003,30(12):2513-2516.
[3]Henter JI,Horne A,Arico M,et al.HLH-2004:diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2007,48(2):124-131.
[4]胡坚,黄文玉,张咏梅,等.幼年类风湿性关节炎合并巨噬细胞活化综合征2例报告[J].中国实用儿科杂志,2001,16(4):247-248.
[5]胡坚,李崇巍,马继军,等.儿童风湿性疾病合并巨噬细胞活化综合征[J].中华儿科杂志,2006,44(11):818-823.
[6]李永柏,胡坚.巨噬细胞活化综合征专题讨论会纪要[J].中华儿科杂志,2006,44(11):831-832.
[7]胡坚.儿童期系统性红斑狼疮与巨噬细胞活化综合征[J].实用临床儿科杂志,2007,25(9):736-749.
[8]Frosch M,Roth J.New insights in systemic juvenile idiopathic arthritis—from pathophysiology to treatment[J].Rheumatology(Oxford),2008,47(2):121-125.
[9]Deane S,Selmi C,Teuber SS,et al.Macrophage activation syndrome in autoimmune disease[J].Int Arch Allergy Immunol,2010,153(2):109-120.
[10]Pamuk ON,Pamuk GE,Usta U,et al.Hemophagocytic syndrome in one patient with adult-onset Still’s disease.Presentation with febrile neutropenia[J].Clin Rheumatol,2007,26(5):797-800.
[11]Filipovich AH.Hemophagocytic lymphohistiocytosis(HLH)and related disorders[J].Hematology Am Soc Hematol Educ Program,2009:127-131.
[12]Billiau AD,Roskams T,Van Damme-Lombaerts R,et al.Macrophage activation syndrome:characteristic findings on liver biopsy illustrating the key role of activated,IFN-gamma-producing lymphocytes and IL-6-and TNF-alpha-producing macrophages[J].Blood,2005,105(4):1648-1651.
[13]Behrens EM,Beukelman T,Gallo L,et al.Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis:data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry(PASOJAR)[J].J Rheumatol,2008,35(2):343-348.
[14]Behrens EM,Canna SW,Slade K,et al.Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice[J].J Clin Invest,2011,121(6):2264-2277.
[15]Brunner HI,Huggins J,Klein-Gitelman MS.Pediatric SLE—towards a comprehensive management plan[J].Nat Rev Rheumatol,2011,7(4):225-233.
[16]Yoshiyama M,Kounami S,Nakayama K,et al.Clinical assessment of Mycoplasma pneumoniae-associated hemophagocytic lymphohistiocytosis[J].Pediatr Int,2008,50(4):432-435.



