路 灿 徐惠君 贾勇圣 佟仲生
受体相互作用蛋白3(receptor-interacting pro⁃ tein 3,RIP3)是受体相互作用蛋白家族的一员,是肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)介导的细胞凋亡和细胞坏死之间的分子开关,在凋亡受到抑制时,RIP3可以通过调节能量代谢而介导细胞坏死[1]。它广泛表达于胚胎和大量成熟组织中,但在一些肿瘤细胞中低表达,如宫颈癌细胞HeLa,骨肉瘤细胞U2OS等[2]。本研究拟构建RIP3基因的真核表达载体,建立稳定过表达RIP3基因的MCF7细胞系,并证实其融合蛋白在细胞内的表达、定位及介导坏死的功能,为进一步探讨程序性坏死的分子机制奠定基础。
1 材料与方法
1.1 主要试剂 Phusion Hot StartⅡDNA polymerase、Pierce BCA Protein Assay Kit蛋白浓度测定试剂盒购自Thermo公司;限制性内切酶NheⅠ、NotⅠ购自Fermentas公司;琼脂糖凝胶回收试剂盒,In-fusion连接酶,E.coliDH5α菌株,质粒提取试剂盒均购自Takara公司;Dulbecco’s Modi fi ed Eagle’s Medium(DMEM)培养液购自neuron-biotech公司;胎牛血清(FBS)购自兰州民海生物工程有限公司;青链霉素(PS)混合液购自北京Solarbio Science and Technology公司;0.25%胰酶购自天津百若克医药生物技术有限公司;聚凝胺、杀稻瘟菌素均购自Santa Cruz公司,RIP3鼠兔源单克隆抗体购自天津三箭生物技术有限公司。pCDH-CMV-MCS-EF1-blastici⁃din-mCherry载体为本实验室已有资源,经转化E.coliDH5α菌株扩增后,用DNA/RNA定量仪测定DNA浓度,置-20℃冰箱保存。
1.2 细胞培养 MCF7、MDA-MB-231、T47D、MDA-MB-435、MCF10A、293T细胞系为本实验室保存。MCF10A细胞用DMEM/F12培养基(含5%马血清、100 μg/L霍乱毒素、10 mg/L胰岛素、20 μg/L表皮生长因子、1.4×106mol/L氢化可的松、1%青链霉素),其余细胞用DMEM培养基(含10%胎牛血清、1%青链霉素)在37℃5%CO2培养箱中培养。
1.3 RT-PCR检测乳腺癌细胞及正常乳腺上皮细胞中RIP3 mRNA的表达 分别收集乳腺癌细胞株MCF7、MDA-MB-231、T47D、MDA-MB-435及正常乳腺上皮细胞MCF 10A各约1×106个,按Trizol和反转录酶试剂说明书分别提取RNA,用分光光度计测定浓度之后,用逆转录试剂盒得到cDNA,以GAPDH作为内参照,进行PCR扩增。引物序列:RIP3上游5'-GAGTTGCCAACCGAACCATCACT-3',下游5'-TACCGTG⁃GAGACAGCATTCA-3'。PCR反应体系20 μL,反应条件:预变性95℃3 min,变性95℃10 s,退火65.5℃30 s,延伸72℃30 s,共35个循环,72℃5 min后,温度降至4℃。扩增长度为243 bp。反应产物于1%琼脂糖凝胶进行电泳,UVP扫描并分析结果。
1.4 RIP3全长编码序列PCR扩增 以逆转录得到的MCF10A cDNA为模板进行RIP3基因cDNA全长的PCR扩增,上游引物:5'-TGTACAAGTCTAGAGCTAGCATGTCGTGC⁃GTCAAGTTA-3',下游引物:5'-CGCGGCCGCGGATCCT⁃TATTTCCCGCTATGATT-3',包含NheⅠ、NotⅠ酶切位点。PCR反应体系为50 μL,含模板50 ng,10×Phusion缓冲液5 μL,dNTPs每种200 μmol/L,引物0.5 μmol/L和Phusion DNA聚合酶0.5 μL。PCR反应条件为:预变性98℃30 s,变性98 ℃ 10 s,退火62 ℃ 30 s,延伸72 ℃ 30 s,20个循环,72 ℃10 min后,温度降至4℃。扩增长度为1 587 bp。扩增产物经1%琼脂糖凝胶电泳后,用凝胶回收试剂盒回收扩增片段。
1.5 mCherry-RIP3表达载体的构建 将上述扩增回收产物即目的基因与事先用NheⅠ、NotⅠ酶切的pCDH-mCherry载体连接,反应体系:载体1 μL,PCR回收产物3 μL,In-fusion连接酶2.5~3 μL,加入H2O共15 μL,室温连接30 min后,将连接反应混合物转化E.coliDH5α,经氨苄霉素筛选,挑取单个克隆培养后,取菌液用小提试剂盒提取质粒,NheⅠ、NotⅠ酶切鉴定。鉴定产物用0.8%琼脂糖凝胶电泳并在凝胶成像分析仪上成像。鉴定正确的重组载体送基因公司测序,经测序比对正确后,再大量扩增。
1.6 包被慢病毒建立稳定表达mCherry-RIP3的MCF7细胞株 转染前1 d铺种293T细胞于6 cm细胞培养皿中,使细胞汇合度达到50%。取20 μg构建的mCherry-RIP3表达载体,两种包装载体各10 μg,钙法转染293T细胞。同时转染不含目的基因的荧光载体作为对照。培养48 h后离心收集上清,-80℃贮存。感染前1 d铺种MCF7细胞于6孔板中,每孔2×105个细胞。感染时,弃去细胞培养液,加入600 μL病毒液及1 μL的聚凝胺,继续培养48 h后,观察荧光,重悬细胞并加入4 mg/L的杀稻瘟菌素药筛,维持药物浓度1周,分别可得到稳定表达RIP3的细胞株及空白荧光载体的细胞株。
1.7 Western Blot 将MCF7细胞、构建的表达mCherry-RIP3及空载体的MCF7细胞铺种于6孔板中,培养至汇合度为60%~80%后,每孔加入200 μL细胞裂解液(Lysis Buffer)裂解MCF7细胞,收集蛋白。各样品取30 μg蛋白上样电泳,转硝酸纤维素膜(Nitrocellulose Blotting Membranes,NC)过夜。第2天将NC膜转移至封闭液(5%脱脂奶粉)中,摇床封闭1 h,孵育Anti-RIP3兔源的单克隆一抗1 h,TBS-T冲洗,孵育荧光标记的二抗1 h,TBS-T冲洗,红外双色荧光扫描成像仪,观察结果。
1.8 TNF-α及Caspase抑制剂Z-VAD-FMK处理下MCF7的死亡情况 将表达mCherry-RIP3及空载体的MCF7细胞(细胞数3×105个/孔)分别铺于6孔细胞培养板(每种细胞各3个孔)过夜,分别给予 0 mg/L、50 mg/L TNF-α及20 μmol/L Z-VAD-FMK预处理30 min,再加入TNF-α至终浓度50 mg/L处理12 h后,显微镜下观察细胞形态,每种处理随机取3个视野计算细胞死亡数及细胞总数。之后在细胞培养液中加入Hoechst 33342染料染色10 min,荧光显微镜下观察细胞核的形态。
1.9 统计学方法 实验数据采用SPSS 17.0统计软件分析,数据以±s表示,各组间比较采用t检验,每组内不同处理方式比较采用方差分析,P<0.05为差异有统计学意义。
2 结果
2.1 乳腺癌细胞系RIP3 mRNA的表达 RIP3 mRNA在MCF10A中表达较高,而在4种乳腺癌细胞系中普遍低表达,其中MDA-MB-435和MDA-MB-231细胞相对较高,T47D和MCF7细胞的表达量相对较低,见图1。本实验选择MCF7细胞用于转染RIP3重组质粒的研究。
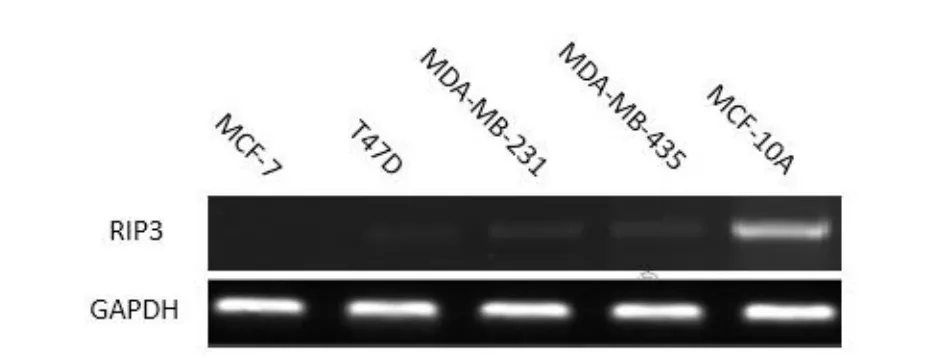
Figure 1 Different mRNA levels of RIP3 in 4 kinds of breast cancer cells and 1 kind of mammary epithelial cell MCF10A图1 RIP3 mRNA在4种不同乳腺癌细胞及正常乳腺上皮细胞MCF10A内的表达
2.2 RIP3基因全长的PCR扩增及载体的酶切 以cDNA为模板,PCR产物经0.8%琼脂糖凝胶电泳显示,扩增产物约1 587 bp,与预期大小一致。同时用限制性内切酶NheⅠ、NotⅠ酶切pCDH-mCherry载体,见图2。
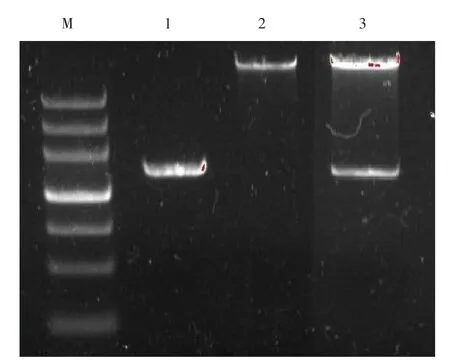
Figure 2 PCR amplification product of RIP3 cDNA and restriction enzyme digestion of the recombinant plasmid mCherry-RIP3图2 RIP3基因全长的PCR扩增产物及重组载体的双酶切鉴定
2.3 RIP3重组载体的鉴定及其序列的测定 将RIP3重组载体用NheⅠ、NotⅠ酶切,可见切出大小约1 560 bp的条带,见图2。测序结果经Pubmed Blast比对后证实插入pCDH-mCherry载体的目的基因正确无误。
2.4 mCherry-RIP3融合蛋白在MCF7细胞中的表达 将mCherry-RIP3基因通过慢病毒导入MCF7细胞中,经Western blot检测到了mCherry-RIP3融合蛋白的表达,分子质量约为85 ku,见图3A。
2.5 mCherry-RIP3融合蛋白在细胞中的定位 通过荧光显微镜观察到表达mCherry-RIP3的MCF7只有细胞质中可见红色荧光分布,见图3B。
2.6 mCherry-RIP3融合蛋白在MCF7细胞中介导TNF-α诱导的程序性坏死 表达mCherry-RIP3及空载体的MCF7细胞经TNF-α处理均有约50%的细胞表现出凋亡的形态特征:细胞缩小变圆,胞膜清晰完整,核固缩。而Z-VAD-FMK预处理后再用TNF-α处理,转入空载体的MCF7细胞的凋亡被抑制,而表达mCherry-RIP3的MCF7细胞死亡数目增加且表现出明显的坏死样形态特征:形态不规则,胞膜模糊,核碎裂溶解,见图4。不同处理方法的空载体-MCF7细胞和RIP3-MCF7细胞的存活率均不相同,差异有统计学意义(均P<0.01)。未处理及TNF-α处理下的空载体-MCF7与RIP3-MCF7细胞的存活率差异无统计学意义,而在TNF-α联合Z-VADFMK处理下,RIP3-MCF7细胞的存活率明显低于空载体-MCF7的细胞存活率(P<0.01),见表1。

Table 1 The cell survival rate of RIP3/Vector-MCF7 under the treatment of TNF-α and Z-VAD-FMK表1 TNF-α及Z-VAD-FMK处理下RIP3/空载体-MCF7细胞的存活率 (n=3,%)
3 讨论
近年来,程序性坏死成为继凋亡之后细胞死亡方式的又一研究热点。传统理论认为,坏死是一种细胞在受到一些物理因素、化学因素或生物因素等环境因素的伤害时出现的大量急速死亡,是一种被动的死亡[3]。后来人们发现TNF-α不但能诱导多种细胞的凋亡,而且还能诱导小鼠成纤维细胞L929的坏死[4],并且Caspase抑制剂Z-VAD-FMK能加强TNF-α诱导的L929细胞坏死[5]。这表明这种细胞坏死可以由特定因子启动,按照一定通路和程序发生,并且是可控的,因而称为程序性坏死[6]。韩家淮教授课题组研究发现,在低表达RIP3的NIH3T3细胞中,TNF-α诱导的凋亡可以被caspase的广泛抑制剂ZVAD-FMK.fmk所抑制;而在高表达RIP3的NIH3T3细胞,Z-VAD-FMK.fmk却可以增强TNF-α诱导的死亡,并且死亡呈现坏死的形态,进而证明了RIP3是TNF-α诱导的细胞凋亡和坏死相互转换的分子开关[7]。异位表达RIP3使得对TNF-α诱导性细胞坏死有抗性的细胞转变成敏感型细胞[2]。Sakon等[8]证明TNF-α诱导产生的活性氧簇(ROS)积聚介导了小鼠胚胎成纤维(MEF)细胞的坏死性死亡。对其具体的执行机制研究表明,RIP3活化后,通过激活糖原磷酸化酶(glycogen phosphory lase,PYGL)、谷氨酸氨基连接酶(glutamate amino ligase,GLUL)和谷氨酸脱氢酶1(glutamate dehydrogenase 1,GLUD1)产生ROS积聚导致细胞坏死[9]。
更深入的研究RIP3基因的功能及其下游的相互作用因子将有助于人们发现新的治疗靶点。基因功能的研究需借助转基因技术,本实验通过从cDNA中PCR扩增出RIP3基因全长,连接至含有红色荧光蛋白mCherry的真核表达慢病毒载体中,联合利用酶切鉴定以及基因测序分析的方法,证实了mCher⁃ry-RIP3重组质粒构建成功。用慢病毒感染本身低表达RIP3的MCF7细胞,构建稳定细胞株;用West⁃ern技术证明其高效表达mCherry-RIP3;在荧光显微镜下观察其转染效率及融合蛋白的细胞定位[10],并通过TNF-α和Z-VAD-FMK的处理证明了其在MCF7细胞中介导坏死的作用。
程序性坏死参与多种疾病的发生发展,如脑缺血和糖尿病等代谢疾病,神经退行性疾病,肿瘤等[11-12]。程序性坏死在肿瘤细胞中作为一种凋亡的替代死亡方式,其功能缺陷或相关基因突变可能参与某些肿瘤的发生发展[13]。有研究表明程序性坏死不但可以加速肿瘤细胞的死亡还可能增加肿瘤细胞对抗癌药物的敏感性[14-15]。本研究通过构建mCherry-RIP3融合蛋白真核表达载体,建立稳定高表达RIP3的细胞系,可以实时观察RIP3的功能定位及其对细胞在不同刺激下凋亡、坏死等死亡机制的影响,以及RIP3调节的下游靶基因的启动子、mRNA及蛋白水平的表达,便于进一步研究其在程序性坏死信号转导中所担当的角色,从而更有效地调控细胞的死亡。为缺血再灌注损伤,神经退行性变,肿瘤的治疗提供新的靶点[16-17]。

Figure 3 Western blotting of mCherry-RIP3 fusion protein using anti-RIP3 antibody(A)and cellular localization(B)图3 mCherry-RIP3融合蛋白的Western blot鉴定(A)与细胞定位(B)
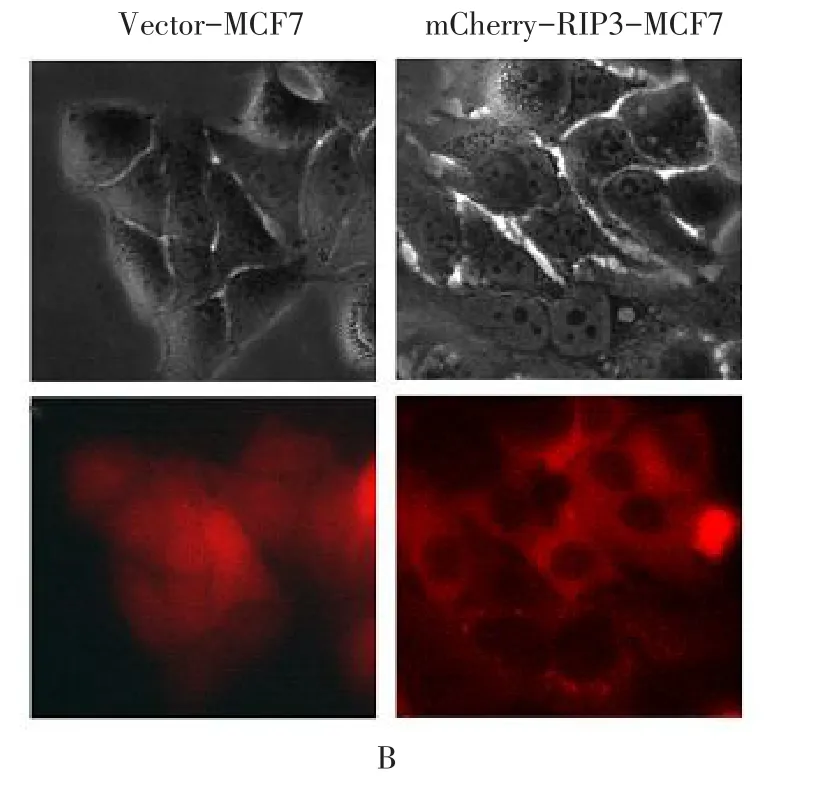
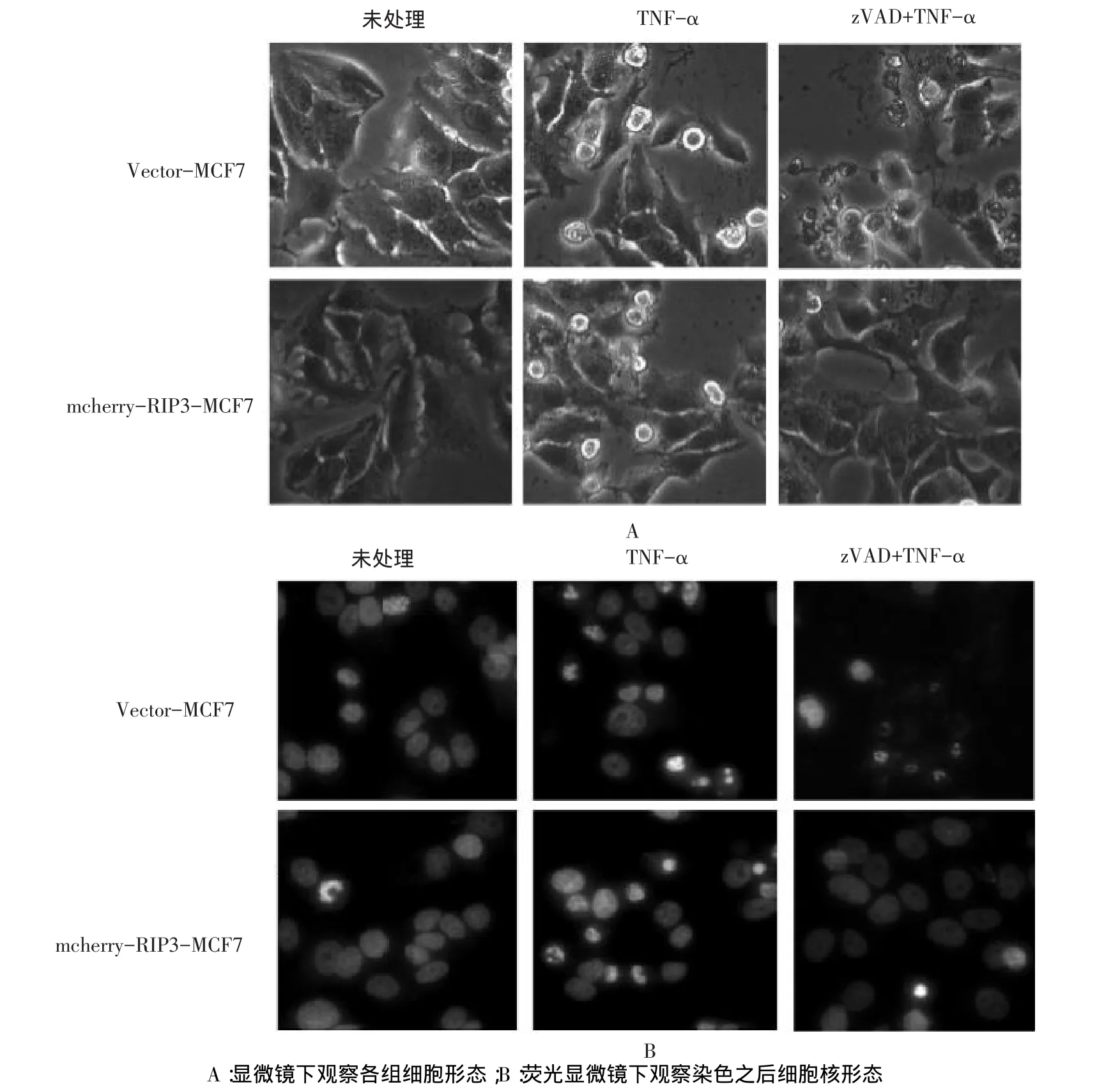
Figure 4 The increased sensitivity of mCherry-RIP3 transfected MCF7 cells to TNF-α and Z-VAD-FMK induced cell necrosis图4 mCherry-RIP3转染后增强MCF7细胞对TNF-α联合Z-VAD-FMK诱导的细胞坏死的敏感性
[1]Zhang D,Lin J,Han J,et al.Receptor-interacting protein(RIP)ki⁃nase family[J].Cell Mol Immunol,2010,7(4):243-249.
[2]He S,Wang L,Miao L,et al.Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha[J].Cell,2009,137(6):1100-1111.
[3]Vandenabeele P,Declercq W,Vanden Berghe T.Necrotic cell death and'necrostatins':now we can control cellular explosion[J].Trends Biochem Sci,2008,33(8):352-355.
[4]Andreas L,Jan HB,Federica DZ,et al.Dichotomy between RIP1-and RIP3-mediate necroptosis in tumor necrosis factor-α-induced shock[J].Molecular Medicine,2012,18(5):577-586.
[5]Wu Y,Tan H,Huang Q,et al.Z-VAD-FMK-induced necroptosis in L929 cells depends on autocrine production of TNF alpha mediat⁃ed by the PKC-MAPKs-AP-1 pathway[J].Cell Death Differ,2011,18(1):26-37.
[6]Degterev A,Huang Z,Boyce M,et al.Chemical inhibitor of nonapop⁃totic cell death with therapeutic potential for ischemic brain injury[J].Nat Chem Biol,2005,1(2):112-119.
[7]Zhang D,Tan H,Huang Q,et al.RIP3,an energy metabolism regu⁃lator that switches TNF-induced cell death from apoptosis to necro⁃sis[J].Science,2009,325(5938):332-336.
[8]Sakon S,Xue X,Takekawa M,et al.NF-kappaB inhibits TNF-in⁃duced accumulation of ROS that mediate prolonged MAPK activa⁃tion and necrotic cell death[J].EMBO J,2003,22(15):3898-3909.
[9]Vandenabeele P,Declercq W,Van Herreweghe F,et al.The role of the kinases RIP1 and RIP3 in TNF-induced necrosis[J].Sci Signal,2010,3(115):re4.
[10]Yang Y,Ma J,Chen Y,et al.Nucleocytoplasmic shuttling of recep⁃tor-interacting protein 3(RIP3):identification of novel nuclear ex⁃port and import signals in RIP3[J].J Biol Chem,2004,279(37):38820-38829.
[11]Dunal Z,Bauer PI,Mihalik R.Necroptosis:biochemical,physiologi⁃cal and pathological aspects[J].Pathol Oncol Res,2011,17(4):791-800.
[12]Bonapace L,Davidson S,Lim S,et al.Induction of autophagy-de⁃pendent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance[J].J Clin In⁃vest,2010,120(4):1310-1323.
[13]Alameda JP,Moreno-Maldonado R,Navarro M,et al.An inactivat⁃ing CYLD mutation promotes skin tumor progression by conferring enhanced proliferative,survival and angiogenic properties to epider-mal cancer cells[J].Oncogene,2010,29(50):6522-6532.
[14]Bonapace L,Bornhauser BC,Schmitz M,et al.Induction of autopha⁃gy-dependent necroptosis is required for childhood acute lympho⁃blastic leukemia cells to overcome glucocorticoid resistance[J].J Clin Invest,2010,120(4):1310-1323.
[15]Mantel F,Frey B,Haslinger S,et al.Combination of lonising irradiation and hyperthermia activates programmed apoptotic and necrot⁃ic cell death pathways in human colorectal carcinoma cells[J].Strahlenther Onkol,2010,186(11):587-599.
[16]Meloni BP,Meade AJ,Kitikomolsuk D,et al.Chracterisation of neu⁃ronal cell death in acute and delayed in vitro ischemia(oxygen-glu⁃cose deprivation)models[J].J Neurosci Methods,2011,195(1):67-74.
[17]Yu X,Deng Q,Bode AM,et al.The role of necroptosis,an alterna⁃tive form of cell death,in cancer therapy[J].Expert Rev Anticancer Ther,2013,13(7):883-893.



