宋咏刚,余鹏 ,胡文慧,石玉玲,赵雪,胡祖权,王赟,曾柱,3**
(1.贵州医科大学 基础医学院,贵州 贵阳 550025;2.贵州医科大学 贵州省免疫细胞与抗体工程研究中心 生物与医学工程重点实验室,贵州 贵阳 550025;3.贵州医科大学 生物与工程学院,贵州 贵阳 550025)
前列腺癌是全球第二大男性癌症,同时也是全球第四大癌症,有着极高的发病率和死亡率[1]。前列腺癌是一种无症状的肿瘤,在整个癌症发生过程中没有任何症状,通过临床手段很难检测。前列腺癌不断增长时,会伴随膀胱颈梗阻、癌细胞转移等症状,这时通常不可能进行治愈性治疗[2]。因此,关于前列腺癌的肿瘤标志物的研究至关重要要。miRNA和lncRNA是基因表达调控的重要组成成分,特别是在癌细胞中表达显着,它能帮助理解疾病发生的分子机制[3]。前列腺癌作为一种遗传性疾病,国内外对其基因水平的调控机制和新分子靶点的研究日益增加[4],例如基于自身免疫疗法的Sipuleucel-T前列腺癌疫苗,通过将VISTA或PARP等分子体外激活树突状细胞(dendritic cells,DCs)、将激活的细胞回输到体内产生免疫应答而治疗癌症[5]。本研究从生物信息学角度出发,通过将基因表达数据库(gene expreesion omnibus,GEO)与癌症和肿瘤图谱中心(the cancer genome atlas,TCGA)数据库关于前列腺癌的样本数据相结合,筛选前列腺癌发生过程中的关键基因及其上下游的调控分子,为临床诊断和临床靶向药物的研发提供理论基础。
1 资料与方法
1.1 基因芯片数据的获取
登录美国国家生物技术信息中心(national centerfor for biotechnology information,NCBI)的GEO数据库(http://www.ncbi.nlm.nih.gov/,GEO database),检索Affymetrix芯片数据GSE55945数据包含13例前列腺癌组织和8例正常组织的样本数据;登录TCGA数据库检索前列腺癌的转录组数据并下载,包含504例前列腺癌组织和55例癌旁组织的样本数据。
1.2 差异基因的分析
1.2.1差异基因的分析 使用RStudio软件‘affy’包对GEO原始数据文件进行标准化处理,并检验归一化处理的结果。使用‘limma’包[6]筛选表达有显着差异的基因(differentially expressed genes,DEGenes),以|logFC|≥2(fold change,FC)并且P<0.05 作为标准进行筛选[7]。通过RStudio软件‘edgR’包对TCGA的前列腺癌数据进行分析[8],同样以|logFC|≥2并且P<0.05作为标准进行筛选。
1.2.2DEGenes的GO和KEGG富集分析和蛋白质相互作用网络的构建 登录KOBAS(http://kobas.cbi.pku.edu.cn/):对筛选出的DEGenes的进行GO分析和京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes ,KEGG)途径分析[9];登录STRING在线工具(https://string-db.org/)[10]:对筛选出的DEGenes构建蛋白质相互作用网络(protein-protein interaction network,PPI)[11]。将结果导入CYTOSCAP软件,使用‘MCODE’插件[12]对该相互作用网络进行K 核解析(k-core decomposition),筛选出DEGenes的核心模块。筛选条件为P<0.05并且基因数目>5[13]。
1.2.3核心基因相应miRNA的预测 将筛选出的核心DEGenes导入mirDIP数据库,对核心DEGenes的miRNA进行预测[14]。
1.2.4miRNA的生存分析及lncRNA的预测 登录STARBASE(http://starbase.sysu.edu.cn/),使用Pan-Cancer工具对预测出的miRNA进行生存分析,选择与前列腺癌患者生存率相关(P<0.05)的miRNA作为核心miRNA。对符合条件的miRNA进行下游lncRNA的预测,为提高预测结果的准确性,选择至少在一种以上的肿瘤中被证实(P<0.05)的lncRNA作为结果。
1.2.5基因mRNA-pathwang网络的构建 将符合标准的mRNA、miRNA、lncRNA以及重要的信号通路及之间的关系导入CYTOSCAPE软件中进行可视化,建立mRNA-pathwang网络图。
2 结果
2.1 由 RStudio软件获得DEGenes
芯片数据(GSE55945)经分析获得正常组织相对于前列腺癌组织的差异表达基因共431个(|logFC|≥2,P<0.05)。TCGA数据库关于前列腺癌的样本经分析,获得癌旁组织相对于前列腺癌组织的差异表达基因共2 472个(|logFC|≥2,P<0.05),部分差异表达基因及其对应信息见表1与表2。利用RStudio软件‘venn’包对两个数据库中筛选出的差异基因使用Merge函数,得到两个数据库共同的137个DEGenes,并构建维恩图(图1)。
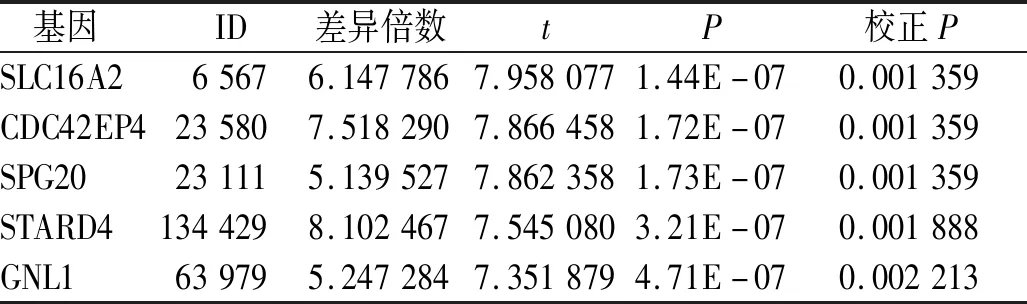
表1 GEO数据库正常组织与前列腺癌组织的部分差异表达基因Tab.1 The partial DEGenes of prostate cancer tissue to normal tissue in GEO database
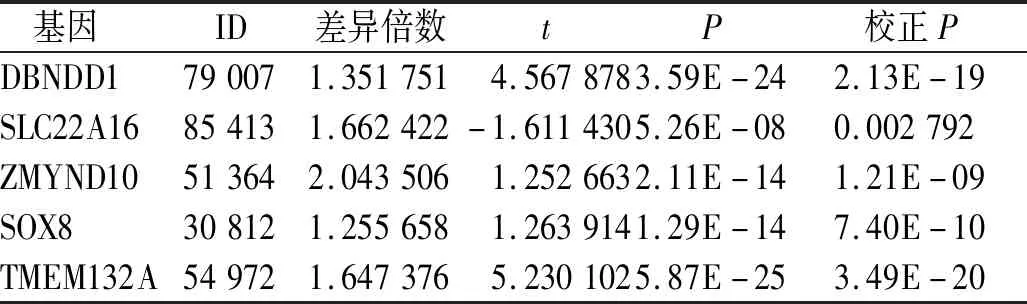
表2 TCGA数据库中正常组织与前列腺癌组织的部分差异表达基因Tab.2 The partial DEGenes of prostate cancer tissue to normal tissue in TCGA database
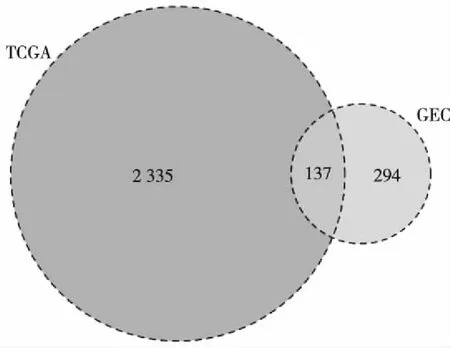
图1 共同差异表达基因的维恩图Fig.1 Venn diagram of common DEGenes
2.2 GO和KEGG富集分析DEGenes
使用KOBAS网站对筛选出的137个DEGenes进行GO分析和KEGG分析,GO分析结果显示有23个GO富集(表3),KEGG富集分析有20个通路被富集(表4)。
2.3 构建DEGenes 的PPI和核心基因相应miRNA的预测
通过STRING在线分析工具绘制DEGenes的PPI(图 2),结果用CYTOSCAPE软件的“MCODE”工具对该PPI进行K 核解析,以节点数>5、所得分数>0.4筛选出子网络,对其中最稳定的子网络作为核心模块进行分析,核心模块包含7个节点基因和 21条相互作用关系对(图 3)。使用mirDIO数据库对7个核心基因预测得到12个对应的miRNA,通过CYTOSCAPE软件对结果进行可视化(图3)。
2.4 miRNA的生存分析及lncRNA的预测
登录STARBASE 网站对预测出的12个miRNA进行生存分析,其中has-let-7a-5p和has-miR-98-5是与前列腺癌患者总体存活率相关的miRNA(P<0.05,图4、表5);通过Pan-Cancer工具验证其对应的基因在前列腺癌患者的表达量,AURKB、BUB1B和EZH2在前列腺癌患者体内高表达(图5)。
2.5 构建lncRNA-mRNA pathway 网络
综合以上结果,使用CYTOSCAPE软件对预测出的lncRNA、与前列腺癌患者生存率有关的miRNA、核心DEGene以及相关通路构建网络图(图6),该网络涉及3个重要的信号通路、3个核心基因、2个miRNA和4个lncRNA。见图6。
3 讨论
近年来随着分子生物学、测序和数据分析技术的发展,生物信息学被广泛地应用在各种肿瘤标志物或治疗靶点的挖掘中,它可以帮助克服癌症的传统疗法中诊断延迟和主观不可靠评估等缺点[15],特别在前列腺癌的诊断中,其黄金标准仍会有20%以上的假阴性率[16]。本研究通过将TCGA数据库和GEO数据库中前列腺癌的样本数据相结合,挖掘出前列腺癌高度相关的差异表达基因137个。对这些基因进行K-核解析后得到7个核心(KIF4A、AURKB、BUB1B、TPX2、NCAPG、HJURP和EZH2)基因,其中BUB1B与前列腺癌的潜在致病风险有关,BUB1B主要参与细胞周期的生物过程,通过与CCNB1、CDK1等基因协同作用,影响前列腺癌患者的疾病进展[17]。SHA等[18]发现PRKAR2B主要加速细胞周期生物学过程并调节多个细胞周期基因,通过过度表达促进趋势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)增殖和侵袭,并抑制CRPC细胞的凋亡。组蛋白甲基转移酶EZH2d的失调,会导致肿瘤抑制性miRNA的表观遗传学沉默而增强前列腺癌细胞增殖和转移过程中的致癌作用[19]。
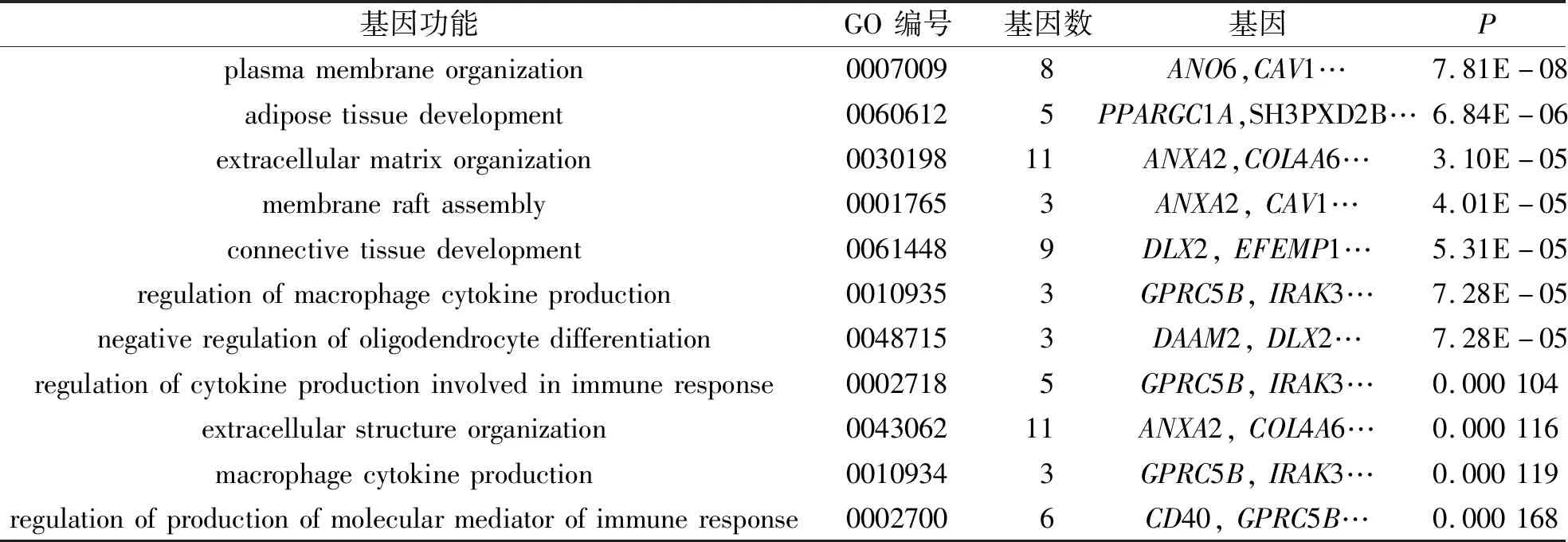
表3 差异表达基因的GO分析Tab.3 GO analysis of DEGenes
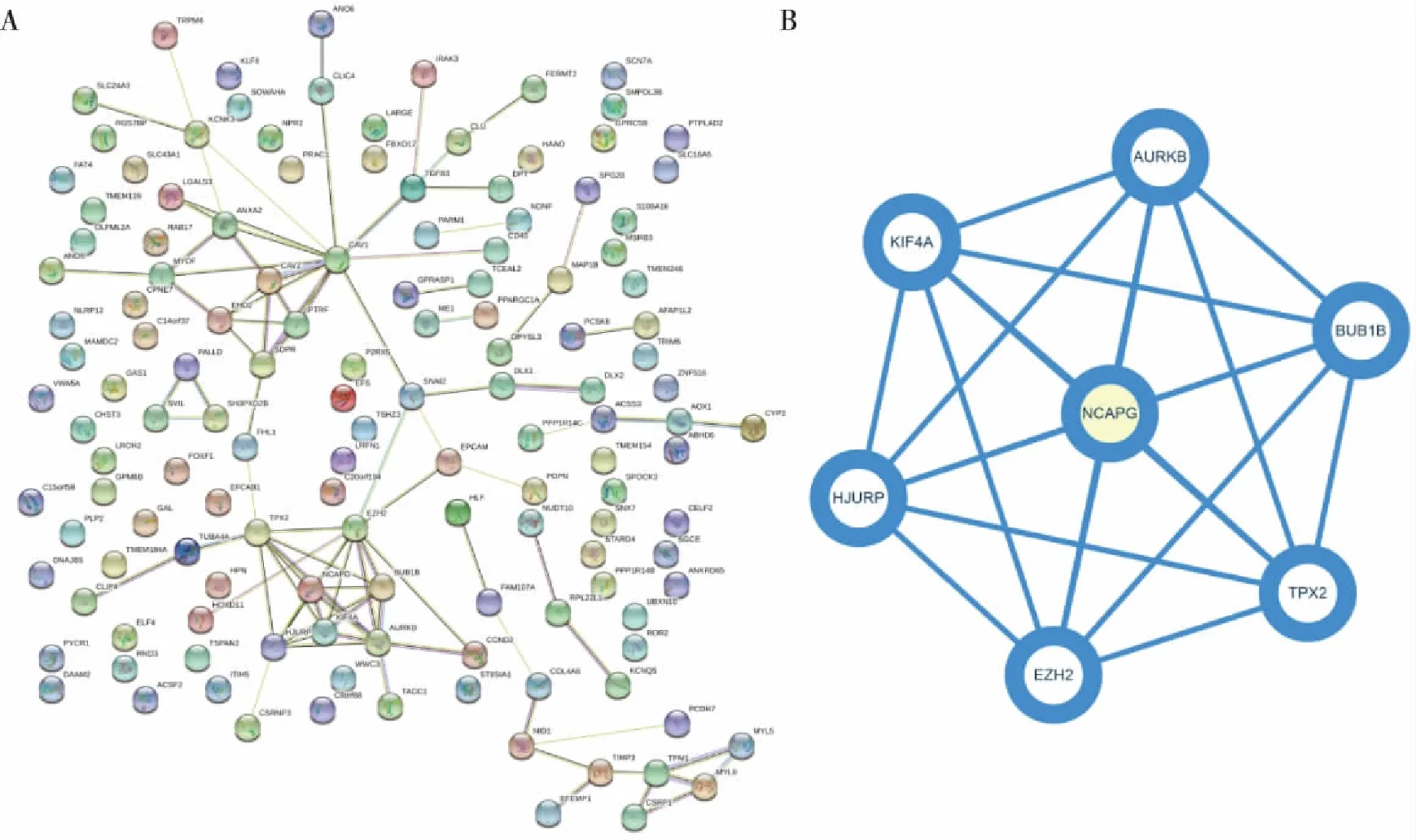
注:A为核心模块,B为核心模块相互作用网络图。图2 差异表达蛋白的核心模块与核心模块相互作用网络Fig.2 The central modules of PPI network of DEGenes constoucted by using CYTOSCAPE and core module
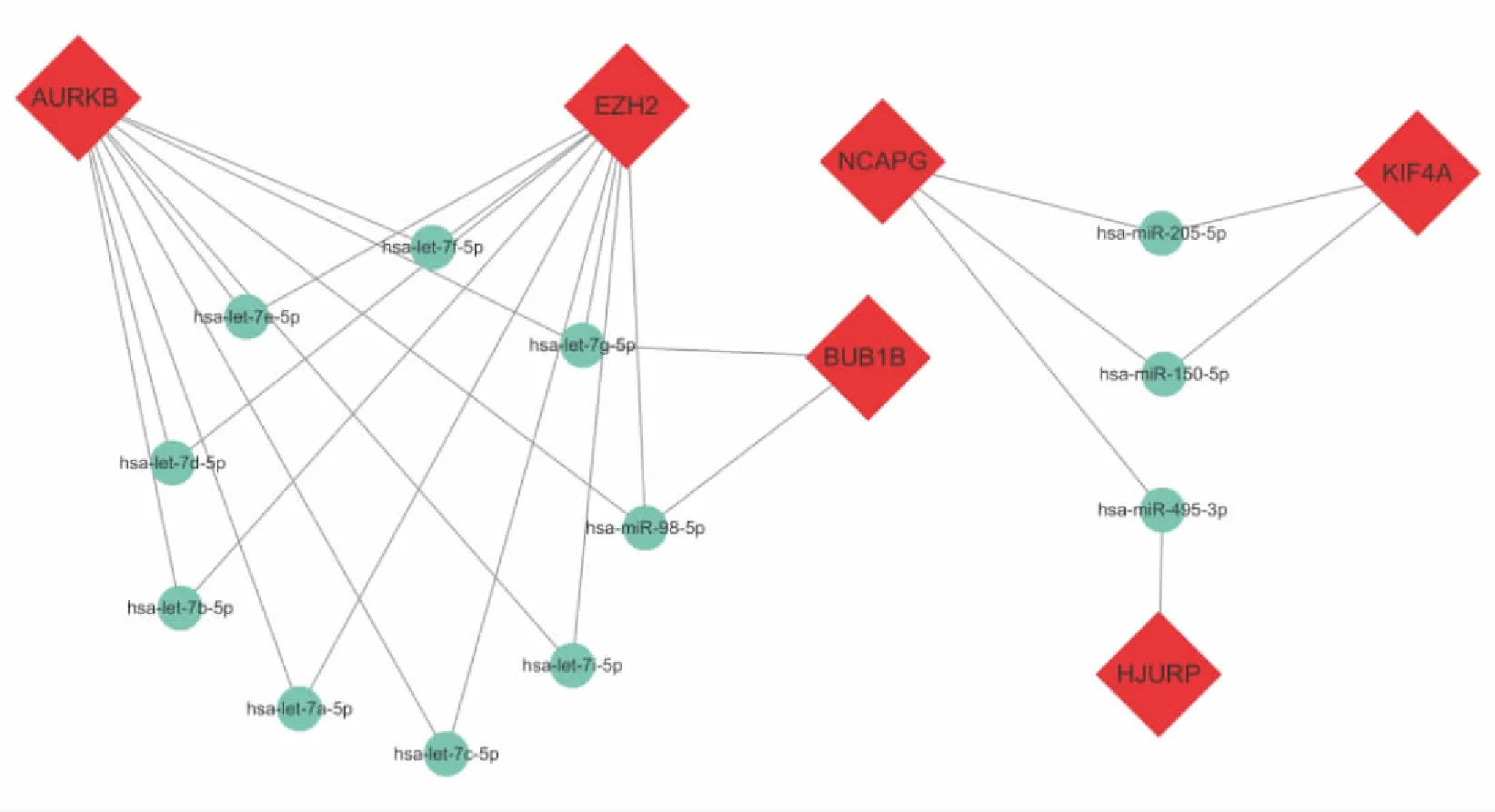
图3 DEGenes 及其对应的miRNAFig.3 DEGenes and corresponding miRNA

注:A为has-miR-21-5p,B为has-miR-98-5p。图4 miRNA的生存分析Fig.4 Survial analysis of miRNA
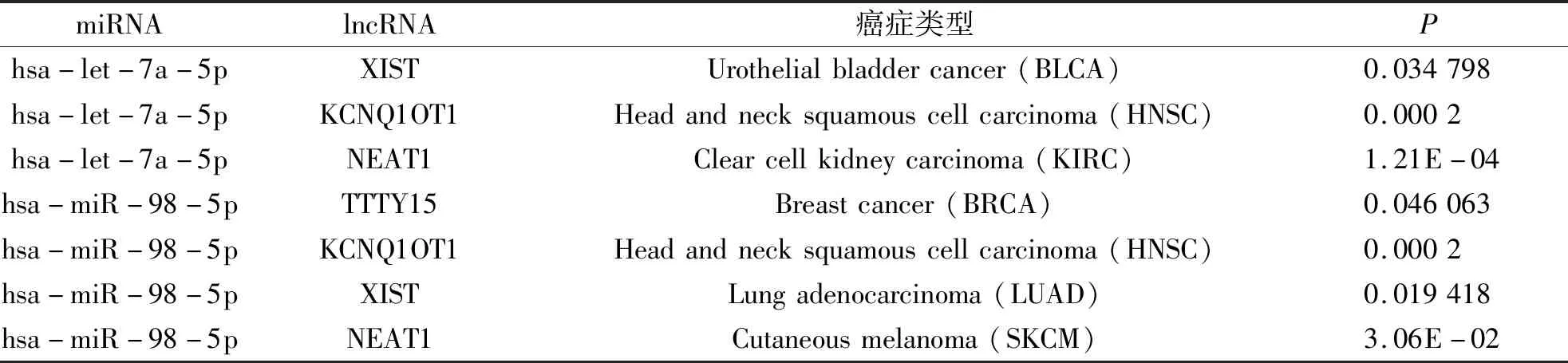
miRNAlncRNA癌症类型Phsa-let-7a-5pXISTUrothelialbladdercancer(BLCA)0.034798hsa-let-7a-5pKCNQ1OT1Headandnecksquamouscellcarcinoma(HNSC)0.0002hsa-let-7a-5pNEAT1Clearcellkidneycarcinoma(KIRC)1.21E-04hsa-miR-98-5pTTTY15Breastcancer(BRCA)0.046063hsa-miR-98-5pKCNQ1OT1Headandnecksquamouscellcarcinoma(HNSC)0.0002hsa-miR-98-5pXISTLungadenocarcinoma(LUAD)0.019418hsa-miR-98-5pNEAT1Cutaneousmelanoma(SKCM)3.06E-02

注:A为BUB1B,B为EZH2,C为MYC。图5 差异基因在前列腺癌中的表达Fig.5 DIfferential expression of DEGenes in prostatic cancer
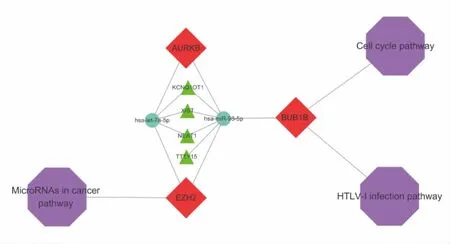
图6 lncRNA-mRNA pathway 网络Fig.6 Network of lncRNA-mRNA pathway
通过对筛选137个DEGenes的GO功能分析和KEGG富集分析发现,这些DEGenes主要涉及细胞粘附,细胞周期和机体炎症反应。CAV1是Src激酶的重要底物,能通过与CSD之间的相互作用抑制粘着斑的形成,引起癌细胞的运动[20]。CD40/CD40L主要参与血管生成,在前列腺肿瘤腺体组织中,CD40在激素难治性期分泌较高,能引起慢性炎症并导致浸润区组织损伤,对前列腺癌进展至关重要[21]。TPM1是一种具有促凋亡功能的肌动蛋白结合肿瘤抑制因子,但在前列腺癌组织中,TPM1发生特异性的可变剪接,通过改变TPM1的结构引起肌动蛋白的失调,导致肿瘤的侵袭和转移,这种作用在肿瘤晚期尤其明显[22]。
非编码RNA在体内不能被翻译为蛋白质,却参与多种细胞过程,主要包括miRNA和lncRNA,由于具有时空或组织特异性,被广泛应用于分子靶点或肿瘤标准物的研究中[23]。本研究通过mirDIP数据库预测核心DEGenes的上游miRNA,并将筛选出的miRNA通过STARBASE数据库中结合临床样本进行生存分析,结果发现hsa-let-7a-5p和hsa-miR-98-5p与前列腺癌患者的总体生存率相关,并且它所对应的3个下游基因AURKB、BUB1B和EZH2在前列腺癌患者体内高表达。其中,hsa-let-7a-5p已被作为I期肺癌的潜在生物标准物之一[24]。hsa-miR-98-5p主要通过下调NEAT1的水平引起非小细胞肺癌的发生发展[25]。对以上2种miRNA进行lncRNA预测,共发现4种lncRNA(XIST、KCNQ1OT1、NEAT1、TTTY15),其中,XIST和KCNQ1OT1能通过促进细胞迁移和上皮-间质细胞转化引起肿瘤的发展[26-27]。TTTY15来定位于Y染色体上,在前列腺癌细胞中高表达,临床研究证实,对TTTY15和USP9Y的测定能预测前列腺癌活检的结果[28]。
本研究从生物信息学角度出发,对前列腺癌的差异基因进行筛选和分析,筛选出前列腺癌发生过程中的关键基因及其上下游的调控分子,构建了一条包含lncRNA、miRNA以及mRNA的分子作用网络,为临床诊断和临床靶向药物的研发提供理论基础。



