高超,杜贵琴,许永劼,朱金凤,潘卫,2,3**,李兴,4
(1.贵州医科大学 医学检验学院,贵州 贵阳 550004;2.贵州医科大学附属医院 贵州省产前诊断中心,贵州 贵阳 550004;3.贵州医科大学 环境污染与疾病监控教育部重点实验室,贵州 贵阳 550004;4.贵州中医药大学 基础医学院,贵州 贵阳 550002)
随着糖尿病发生率的增高,由糖尿病引起的脑代谢异常也日益引起大众的关注[1]。糖尿病脑病是一种多发性因素引起的复杂性病变,胰岛素的绝对或者相对不足、高血糖致使大脑神经元微环境的紊乱是糖尿病脑病发病的重要因素[2-4]。临床研究发现,海马是2型糖尿病患者最先受累的脑区,同时也是阿尔茨海默症最先产生病变的部位[5-6]。海马是记忆巩固的重要部位,发挥着学习以及认知功能的作用,信息储存也是海马组织的功能之一。海马组织属于大脑边缘系统,大部分由椎体神经元构成,种类单一,是体外研究糖尿病脑病细胞模型的理想选择[7]。现阶段,国内外对原代培养海马神经元细胞的方法很多,但都存在一定问题,如胶质细胞过多、细胞纯度过低等。因此,本研究旨在提供一种改良的海马神经元原代培养方法,减少杂质细胞干扰,提高细胞纯度,为后续糖尿病脑病的机制研究提供帮助。
1 材料与方法
1.1材料
1.1.1实验动物和试剂 选取出生24 h内SD乳鼠100只,体质量4~6 g[合格证号为SCXK(黔)2018-0001],由贵州医科大学实验动物中心供给。 试剂包括Neurobasal-A培养基(美国Gibco公司)、DMEM高糖型培养基(美国Gibco公司)、B-27营养因子(美国Gibco公司)、0.25%胰蛋白酶(美国Hyclone公司)、D-hanks液(美国Gibco公司)、胎牛血清(美国Hyclone公司)、马血清(美国Hyclone公司)、左旋多聚赖氨酸(PLL,美国Sigma公司)、L-谷氨酰胺(美国Gibco公司)、CCK-8检测试剂盒(中国凯基公司)、兔抗鼠神经元特异性烯醇化酶(NSE,美国abcam公司)、4%多聚甲醛(索莱宝公司)以及免疫组化抗体稀释液(索莱宝公司)。
1.1.2主要仪器 Thermo-3111型CO2培养箱(日本SANYO公司)、VL-4型无菌操作台(珠海市再鑫仪器有限公司)、IMARK型酶标仪(美国Bio-Rad公司)、倒置显微镜(日本Nikon公司)、数码显微镜(日本Nikon公司)以及超纯水仪(四川沃特尔水处理设备有限公司)。
1.1.3主要溶剂的配置 0.25 mg/L的左旋多聚赖氨酸(PLL)溶液:称取PLL后超纯水溶解,配制0.25 mg/L的溶液,经过滤器过滤细菌后备用。种植型培养基:DMEM培养基、马血清、胎牛血清、L-谷氨酰胺以及双抗,比例为83 ∶10 ∶5 ∶1 ∶1。维持型培养基:Neurobasal-A培养基、B-27、谷氨酰胺以及双抗,比例为96 ∶2 ∶1 ∶1。PBS溶液:称取PBS粉末10 g溶解于1 L超纯水中,高压灭菌备用。
1.2原代培养海马神经元细胞
1.2.1细胞培养板的预处理 取6孔板、24孔板、96孔板进行预处理。其中24孔板预先放置细胞爬片。用0.25 mg/L的左旋多聚赖氨酸(PLL)处理培养板,包被8 h或者过夜,放置在培养箱中,使用前用PBS清洗3遍,置于超净工作台晾干,备用。
1.2.2细胞培养 (1)取出生24 h内SD乳鼠,在75%酒精中清洗30 s,断头后,用弯剪刀剪开头盖骨,将整个大脑放入预冷的D-hanks 液中,在数码显微镜下用眼科镊缓缓将大脑剥开,选取海马体,用镊子轻轻剥去“新月形”海马组织,在数码显微镜下,将海马体上的被膜和血管完全剥离,尽量保证海马的完整性,将剔除干净的海马组织放在预冷的种植培养基中。(2)将取下来的海马组织剪碎至1 mm ×1 mm ×1 mm 大小,静置2 min弃去上清液,加入种植培养基3 mL,用加样枪吹打20次后,静置2 min,吸取上清液,重复该步骤。将所取上清液放入离心机中离心5 min,转速设定850 r/min,把上层液体去掉,37 ℃加入0.125%胰蛋白酶消化20 min,每隔5 min摇晃培养皿,使其充分消化,在显微镜下观察组织块上无细胞后,加入等体积的胎牛血清终止消化,过程中轻轻吹打数次,200目筛网过滤,放入离心机中离心5 min,转速设定850 r/min,把上层液体去掉,制成单细胞悬液加入种植培养基。(3)细胞接种,将6孔板和96孔板事先用L-多聚赖氨酸包被,然后把细胞悬液调整至2×108个/L的浓度种植于其中,培育8 h,倒掉种植培养基,用PBS清洗3遍,换成维持型培养基培养,每隔2 d进行换夜。
1.3海马神经细胞纯度鉴定
将细胞爬片用多聚赖氨酸进行处理,放入24孔培养板中,将细胞悬液以(1~5)×108个/L的浓度接种到其中,放入培养箱培养至5 d,用PBS冲洗细胞爬片3次,每次2 min,后用4%多聚甲醛固定30 min,PBS洗3次,每次5 min。使用NSE免疫组化染色试剂盒鉴定海马神经细胞纯度,随机选取显微镜的视野,重复8次计算100个细胞中海马神经细胞的数量,取其平均值,海马神经元纯度为阳性百分率。
1.4CCK-8法检测细胞活力
将细胞悬液以(1~5)×108个/L的浓度接种到预先经过多聚赖氨酸处理的96孔板中,海马神经元原代培养至第1、3、5、7、9、14天,全部操作严格按照CCK-8试剂盒说明书进行。每组做5个平行孔,培养于37 ℃、5%CO2培养箱内2 d,之后拿出CCK-8溶液,每孔加入10 μL,置于37 ℃水浴箱中孵育2 h,酶标仪测定吸光度(450 nm)。实验重复3次,每次5个副孔。
2 结果
2.1原代海马神经元形态观察
如图1所示,培养第1天的细胞胞体饱满透亮,形状各有不同,呈圆形、椭圆形、不规则形,有光晕明显围绕在细胞周围,且有一部分细胞开始贴壁,细胞上有少量突起,突起之间有少量连接;培养第4天的海马神经元细胞开始呈聚集状态,细胞胞体相对于第1天来说增大,突起较之前更为明显,有突起连接形成网络;培养第4、5及7天的神经元细胞体积变大、成熟饱满,细胞胞浆较丰富,细胞光晕更为明显,突起连接形成的网状结构也更加明显;培养第11、13天的神经元比较成熟,突起连接形成的网状结构也交错复杂,由于一部分神经元细胞开始退化,培养液内有悬浮的退化的细胞碎片使得细胞背景模糊不清晰。

图1 不同培养天数的海马神经元细胞的形态变化(倒置显微镜,×200)Fig.1 Morphological changes of primary hippocampal neurons (inverted microscope,×200)
2.2海马神经元细胞活性检测
培养第1~6天海马神经元细胞活性逐渐增强,第7天时细胞活性达峰值,并在第7~9天有一个短暂的平台期,第11天后细胞活性逐渐下降,见图2。

图2 海马神经元细胞活性检测Fig.2 The viability of cultured primary hippocampal neurons
2.3代海马神经元纯度鉴定
NSE免疫组织化学染色培养7 d细胞,其中部分凸起和胞浆染色为棕灰色或者黄棕色的颗粒,计为海马神经元。海马神经元纯度为90%。见图3。
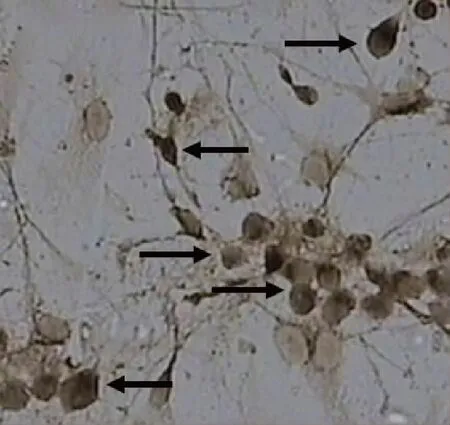
图3 NSE法鉴定海马神经元纯度(NSE,×200)Fig.3 IHC staining of NSE in primary hippocampal neurons(NSE,×200)
3 讨论
在研究神经系统疾病中,神经元是体外细胞培养的理想模型。海马是神经元比较集中的组织,非神经元细胞较少,同时海马作为学习记忆的主要场所,在认知功能中扮演重要角色,故在进行体外神经元的原代培养时作为首选[8-9]。已有研究证明,糖尿病脑病的发生发展与大脑海马区生物学功能的改变密切相关,海马的结构与功能正常与否直接影响到糖尿病患者的认知功能[10]。现阶段,原代培养海马神经元的方法虽多,但培养效果存在很大问题,如杂质细胞过多、背景脏等问题。因此,本研究通过改良传统的培养方法,提供一种稳定高效的海马神经元原代培养方法,能为后续研究糖尿病脑病以及多种神经退行性疾病提供理想的体外细胞模型。
查阅文献[11-12],总结最近几年的原代培养海马神经元方法,发现培养海马神经元主要存在以下几个问题:(1)在取材过程中,不注意海马组织上被膜和血管的剔除。实验结果表明,如果血管未剔除干净,消化分离时会残存较多的红细胞和成纤维细胞,使得在细胞计数和浓度摸索时造成困难,同时也会影响海马神经元本身的生长。被膜中含有较多的神经胶质细胞,胶质细胞本身会竞争性抑制神经元生长发育。本研究在数码显微镜视野下,使用眼科镊将海马组织中的被膜和血管剔除干净,极大降低了杂质细胞。(2)消化不易控制。在取下完整的海马组织后,无法确定最佳的消化工具和作用时间,其中消化工具常为胰酶或木瓜酶。但胰蛋白酶活性强不易控制,任玥等[13]采用木瓜酶,相对于胰蛋白酶力度温和,极大的改进了原代培养中酶消化环节所带来的时间和剂量难以控制的问题。非酶消化法因成本过高,使用较少。本研究综合上述发现,选择稀释后浓度为0.125%胰蛋白酶为工具,发现该浓度的胰蛋白酶作用温和,减低细胞损伤。在消化时间上,本研究选择15~20 min,每隔5 min摇晃培养皿并观察消化情况,及时终止消化,避免损伤细胞。(3)细胞纯化不足。神经元培养最重要的问题就是如何提高其纯度,抑制非神经元细胞生长。很多研究发现在培养基中加入阿糖胞苷可以显着抑制非神经元细胞生长,但这种方法无法很好控制阿糖胞苷的作用时间和作用浓度,很容易导致神经元细胞生长受到影响[14-15]。还有研究发现,采用 B27无血清培养基可以选择性地使神经元生长,而抑制非神经元细胞的生长和增殖[16-18]。研究同样证明了这一点,加入B27的无血清培养基能够较好的抑制非神经元细胞生长,且作为神经元特异性刺激因子,对神经生长作用显着,能够起到纯化神经元的作用。(4)有研究发现初始种植密度对神经元贴壁无影响,但会严重影响轴突和树突的伸展,从而影响细胞间信号的传递。神经元细胞密度过高过低都会直接影响细胞的状态,当密度过大,细胞会出现接触抑制、争夺营养等现象,使其无法完全发育成熟;而过低的培养密度如0.5×108/L也是不利的,Kaech等[19]研究证实,低密度培养下的海马神经元很难维持长时间的培养,发挥生物信息传递的功能。本实验依据李一鹏等[20]方案,选择(1~5)×108个/L细胞浓度,培养出的神经元细胞胞浆丰富、光晕明显,发达的突起向外延伸并且变粗分出许多分支,互相连接形成密集交错的网络,培养至14 d仍有较强活性。
本研究发现,与传统直接将剪碎的组织块加胰蛋白酶消化不同,将海马组织经剪碎、吹打、离心后,易消化得到更纯的神经元。在选材过程中,尽量选择乳鼠,无需解剖孕鼠,获得的神经元细胞活性高,同时避免脑部海马区血管和被膜不易剔除干净的缺点。整个过程中,将脑组织及海马全程浸泡在预冷的D-hanks液或种植培养基中,提高细胞的存活率,降低大脑代谢情况。整个杀鼠过程尽量在3 h内完成,减少组织细胞损伤。经本方法原代培养的海马神经元,细胞胞体形态饱满,细胞表面光晕明显,聚集现象突出,细胞突起粗大且致密,互相交织呈密集的网络结构,经NSE免疫组织化学鉴定纯度高达90%以上,具有很好的实用性。
综上所述,本实验汲取了前人经验教训改良海马神经元原代培养方法,提高了神经元纯度,为体外快速稳定高效培养神经元提供方案,同时也为后续关于糖尿病脑病及其他神经退行性疾病的研究奠定基础。



