黄燕,单丽,邱水生,潘旭东,林雄,罗花彩
作者单位:福建中医药大学附属人民医院,福建 福州350004
益肾降糖胶囊是由我院院内制剂益肾降糖饮(闽药制字Z061060503)剂型改革而得的复方制剂。该方是阮诗玮教授总结古今文献,结合多年临床经验,在中医理论指导下自创的经验方。其处方以制何首乌、赤芍、黄芩、太子参、黄芪、山药、肉苁蓉等14味药材组成,具有滋阴养血、益肾通络之功效。该制剂临床应用于肾气亏虚、阴血亏损、脉络瘀阻等特点的糖尿病肾病,能够有效降低尿微量白蛋白,血浆同型半胱氨酸水平,延缓病情进展的速度,临床应用几十年,效果良好。原制剂质量标准简单,只对方中黄芪、赤芍进行薄层鉴别,无含量测定项目。因剂型由合剂改为胶囊剂,样品处理方法不同,故本研究于2018年9月至2019年12月对原标准中薄层鉴别项目进行整合优化,重新建立了制何首乌、赤芍、玄参、黄芪、山药、黄芩的薄层色谱鉴别。方中制何首乌为君药,具有补肝肾,益精血,乌须发,强筋骨,化浊降脂的作用,其专属有效成分2,3,5,4’-四羟基二苯乙烯-2-0-β-D葡萄糖苷(简称二苯乙烯苷)具有抗氧化、神经保护、肝脏保护、防治老年痴呆等作用,常作为制何首乌质控指标成分。赤芍、黄芩为臣药,赤芍的主要成分芍药苷具有抗自由基损伤、降低血液黏度,抗血小板聚集,改善微循环、抗氧化等作用,黄芩的主要成分黄芩苷具有解热抗炎、清除自由基、抗氧化、心脑血管保护等作用。制何首乌、赤芍、黄芩在益肾降糖胶囊中起至关重要作用,为了完善其质量标准,增加了含量测定项目,采用高效液相色谱法对芍药苷、二苯乙烯苷、黄芩苷同时进行测定,为该制剂质量标准的建立提供一定的参考依据。
1 仪器与试剂
1.1 仪器
HWS26 型电热恒温水浴锅(上海一恒科学仪器有限公司),WD-9403A 型紫外仪(北京市六一仪器厂),98-1-B 型电子调温电热套(天津市泰斯特仪器有限公司),循环水式多用真空泵(郑州长城科工贸有限公司),KQ-500DE 型数控超声波清洗器(昆山市超声仪器有限公司),CPA-225D 型电子分析天平(赛多利斯科学仪器有限公司),Ulit Mate-3000型高效液相色谱仪(戴安中国有限公司)。1.2 试剂
制何首乌对照药材(批次121454-201405),玄参对照药材(批次121008-201609),山药对照药材(批次121137-201603),大黄素对照品(批次110756-200110,含量98.7%),芍药苷对照品(批次110736-201842,含量97.4%),哈巴俄苷对照品(批次111730-201709,含量95.9%),黄芪甲苷对照品(批次110781-201717,含量96.9%),2,3,5,4’-四羟基二苯乙烯-2-0-β-D 葡萄糖苷对照品(批次110844-201713,含量93.6%),黄芩苷对照品(批次110715-201720,含量93.5%),以上均购自中国食品药品检定研究院;硅胶G层析板(批次20180908,青岛海洋化工厂),甲醇(色谱纯,批次20180422国药集团化学试剂有限公司),其余试剂为分析纯;益肾降糖胶囊(批次20181101,20181105,20181108,20181110,0.43 克/粒,福建中医药大学附属人民医院制剂室)。2 方法与结果
2.1 定性鉴别
2.1.1
制何首乌的薄层鉴别 称取益肾降糖胶囊(批次20181101,20181105,20181108)内容物10 g,加2.5 mol/L 硫酸和三氯甲烷各30 mL,摇匀,水浴加热回流40 min,冷却,取三氯甲烷层,水层再用三氯甲烷20 mL振摇提取,三氯甲烷提取液合并,水浴蒸干,用三氯甲烷溶解残渣并定容至1 mL,制成供试品溶液。另称取制何首乌对照药材0.2 g,照上述方法制备对照药材溶液。取不含制何首乌的供试品,同法制备缺制何首乌的阴性对照溶液,再取大黄素对照品适量,加三氯甲烷溶解,制成每1毫升含0.5 mg大黄素的对照品溶液。取上述供试品、缺制何首乌阴性对照溶液各10 μL,对照品和对照药材溶液各5 μL,在同一硅胶G层析板上分别点样,取甲苯—乙酸乙酯—甲酸(体积比为20∶2∶1)的上层溶液置层析缸中,预饱和15 min,薄层板展开至适宜高度,取出,自然晾干,置紫外光灯(365 nm)下观察。薄层色谱中,供试品、对照品、对照药材溶液色谱在薄层板相对应的位置呈同样的黄色斑点,且斑点分离良好,而阴性对照无同样的干扰斑点。该法操作简单,具重现性,可用于方中制何首乌的鉴别。见图1A。2.1.2
赤芍、玄参的薄层鉴别 称取益肾降糖胶囊(批次20181101,20181105,20181108)内容物10 g,加甲醇40 mL,摇匀,超声提取(500 W,40 kHz)30 min,离心,上清液水浴蒸干,用水20 mL 溶解残渣,加正丁醇振摇提取2 次,每次25 mL,合并正丁醇提取液,水浴蒸干,用甲醇溶解残渣并定容至1 mL,制成供试品溶液。另取玄参对照药材1 g,照上述方法制备对照药材溶液。分别取不含赤芍、不含玄参的供试品,同法制备缺赤芍、缺玄参的阴性对照溶液。再取哈巴俄苷、芍药苷对照品适量,分别加甲醇溶解,分别制成每1 毫升含1 mg 哈巴俄苷、芍药苷的对照品溶液。取上述对照品、对照药材溶液各5 μL,供试品及缺玄参、缺赤芍的阴性对照溶液各10 μL,在同一硅胶G 层析板上分别点样,以三氯甲烷—甲醇—水(体积比为12∶4∶1)的下层溶液为展开剂,薄层板展开至适宜高度,取出,自然晾干,5%香草醛硫酸溶液喷雾显色,加热至斑点颜色清晰,置日光灯下检视。薄层色谱中,供试品与对照品和对照药材溶液色谱在相对应的位置呈同样颜色的斑点,且斑点分离良好,而阴性对照溶液无同样的干扰斑点。该法操作简单,具重现性,可用于方中赤芍与玄参的鉴别。见图1B。2.1.3
黄芪的薄层鉴别 称取益肾降糖胶囊(批次20181101,20181105,20181108)内容物15 g,加甲醇30 mL,摇匀,超声提取(500 W,40 kHz)30 min,离心,上清液水浴蒸干,加水20 mL 溶解残渣,用水饱和的正丁醇振摇提取2 次,每次20 mL,合并正丁醇提取液,加氨试液洗涤2次,每次20 mL,弃去氨试液,再用正丁醇饱和的水洗涤2 次,每次30 mL,弃去水液,取正丁醇提取液水浴蒸干,用甲醇溶解残渣并定容至1 mL,制成供试品溶液。另取不含黄芪的供试品,同法制备缺黄芪的阴性对照溶液。再取黄芪甲苷对照品适量,加甲醇溶解,制成每1 毫升含1 mg黄芪甲苷的对照品溶液。取上述3种溶液各10 μL,在同一硅胶G层析板上分别点样,以三氯甲烷—甲醇—水(13∶7∶2,V/V/V
)的下层溶液为展开剂,展开至适宜高度,取出,自然晾干,用10%硫酸乙醇溶液喷雾显色,加热至斑点颜色清晰,置日光灯下观察。薄层色谱中,供试品与对照品溶液色谱在相对应位置呈同样颜色的斑点,且斑点分离良好,而阴性对照溶液无同样颜色的干扰斑点。该法操作简单,具重现性,可用于方中黄芪的鉴别。结果见图1C。2.1.4
山药的薄层鉴别 称取益肾降糖胶囊(批次20181101,20181105,20181108)内容物10 g,加二氯甲烷30 mL,摇匀,回流加热2 h,冷却,离心,上清液水浴蒸干,用二氯甲烷溶解残渣并定容至1 mL,制成供试品溶液。另取山药对照药材2 g,照上述方法制备对照药材溶液。取不含山药的供试品,同法制备缺山药的阴性对照溶液。取上述溶液各10 μL,在同一硅胶G 层析板上分别点样,以三氯甲烷—丙酮(10∶0.5,V/V
)为展开剂,展开至适宜高度,取出,自然晾干,用5%香草醛硫酸溶液喷雾显色,加热至斑点显色清晰,在日光灯下检视。薄层色谱中,供试品与对照药材溶液在相对应位置呈同样颜色斑点,且斑点分离良好,而阴性对照无同样颜色的干扰斑点。该法操作简单,具重现性,可用于方中山药的鉴别。结果见图1D。2.1.5
黄芩的薄层鉴别 称取益肾降糖胶囊(批次20181101,20181105,20181108)内容物10 g,加乙酸乙酯—甲醇(3:1)20 mL,回流加热30 min,冷却,离心,取上清液水浴蒸干,用甲醇溶解残渣并定容至1 mL,制成供试品溶液。另取不含黄芩的供试品,同法制备缺黄芩的阴性对照溶液。再取黄芩苷对照品适量,加甲醇溶解,制成每1 毫升含1 mg 黄芩苷的对照品溶液。取上述溶液各10 μL,在同一硅胶G 层析板上分别点样,以乙酸乙酯—丁酮—甲酸—水(5∶3∶1∶1,V/V/V/V
)为展开剂,展开至适宜高度,取出,自然晾干,用2%三氯化铁乙醇溶液喷雾显色,加热至斑点颜色清晰,在日光灯下观察。薄层色谱中,供试品与对照品溶液在相对应的位置呈同样的暗绿色斑点,且斑点分离良好,而阴性对照无同样颜色的干扰斑点。该法操作简单,具重现性,可用于方中黄芩的鉴别。结果见图1E。
2.2 含量测定(避光操作)
2.2.1
色谱参数的选择 Ultimate® XB-C色谱柱(4.6×250 mm,5 μm);以甲醇(A)-0.2%磷酸溶液(B)为流动相,进行梯度洗脱(0~15 min,30%A,15~20 min,30%~50% A,20~30 min,50% A,30~35 min,50%~30% A);检测波长设置芍药苷为230 nm,二苯乙烯苷和黄芩苷为320 nm;柱温40 ℃;流速为1.0 mL/min;进样量10 μL。在上述色谱条件下,供试品中芍药苷、二苯乙烯苷、黄芩苷与其他成分均分离良好,分离度均>1.5,阴性对照溶液色谱相应位置未显示相同的吸收峰,理论板数以芍药苷、二苯乙烯苷、黄芩苷峰计均不低于3 000,结果见图2。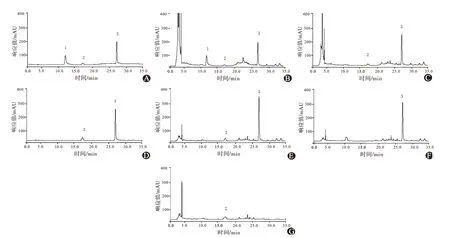
图2 高效液相色谱图:A为对照品溶液(λ=230 nm);B为供试品溶液(λ=230 nm);C为缺赤芍阴性溶液(λ=230 nm);D为对照品溶液(λ=320 nm);E为供试品溶液(λ=320 nm);F为缺首乌阴性溶液(λ=320 nm);G为缺黄芩阴性溶液(λ=320 nm)
2.2.2
对照品混合溶液的配制 分别取芍药苷、黄芩苷、二苯乙烯苷对照品适量,精密称定,分别置棕色容量瓶中,用甲醇溶解并定容至刻度,摇匀,分别制成每1 毫升含芍药苷0.391 5 mg、黄芩苷0.451 0 mg、二苯乙烯苷0.246 2 mg的各对照品贮备液;再精密量取上述芍药苷贮备液5 mL、黄芩苷贮备液5 mL、二苯乙烯苷贮备液1 mL,置同一25 m L 棕色容量瓶中,加甲醇定容,混合均匀,制成每1 毫升含芍药 苷78.300 μg、黄 芩 苷90.200 μg,二 苯 乙 烯 苷9.848 μg的混合对照品溶液。2.2.3
供试品溶液的配制 取益肾降糖胶囊2 粒,倒取内容物混合均匀,取0.34 g,精密称定,置于碘量瓶中,精密加入稀乙醇溶液10 mL,称定重量,超声处理(500 W,40 kHz)30 min,冷却,继续称定重量(用稀乙醇溶液补足减失的重量),摇匀,滤过,取续滤液,即得供试品溶液。2.2.4
阴性对照溶液的配制 分别取不含赤芍、制何首乌、黄芩的阴性样品,按“2.2.3”项下的制备方法分别制成缺赤芍、制何首乌、黄芩的阴性溶液。2.2.5
线性关系考察 分别精密吸取适量的二苯乙烯苷、芍药苷、黄芩苷对照品贮备液,制成每1 毫升含二苯乙烯苷分别为3.939,4.924,7.878,9.848,14.772 μg,含 芍 药 苷 分 别 为31.320,78.300,117.450,156.600,234.900 μg,含 黄 芩 苷36.080,90.200,135.300,180.400,270.600 μg 的5 个系列对照品混合溶液。按“2.2.1”项下设定的色谱参数,上述5个对照品混合溶液各精密吸取10 μL,分别进样进行测定,记录各浓度对照品混合溶液的峰面积。采用Chromeleon 色谱工作站软件进行统计分析,以峰面积(Y^)为纵坐标,对照品质量浓度(X
)为横坐标进行线性回归,计算回归方程,二苯乙烯苷、芍药苷、黄芩苷的回归方程分别为Y
^=0.602 3X
-0.034 0(r
=0.999 5),Y
^= 0.185 2X
+ 0.542 2(r
=0.999 1),Y
^=0.321 0X
+ 0.875 2(r
=0.999 5),结果表明二苯乙烯苷 在3.939 ~14.772 μg/mL,芍 药 苷 在31.320 ~234.900 μg/mL,黄芩苷在36.080 ~270.600 μg/mL范围与峰面积具有良好的线性关系。2.2.6
精密度试验 精密吸取“2.2.2”项下的对照品混合溶液10 μL,按照“2.2.1”项下设定的色谱参数依次进样6次,记录对照品混合溶液的峰面积。结果,芍药苷、黄芩苷、二苯乙烯苷测得峰面积的相对标准偏差(RSD)分别为1.07%,0.92%,0.79%(n
=6)。2.2.7
重复性与稳定性试验 取同一批益肾降糖胶囊(批次20181110),按照“2.2.3”项下的制备方法制备供试品溶液6 份,再分别精密吸取10 μL,按照“2.2.1”项下设定的色谱参数依次进样分析。结果,芍药苷、黄芩苷、二苯乙烯苷平均质量分数分别为4.452 0 mg/g,6.210 5 mg/g,0.225 1 mg/g;RSD 分别为0.48 %,0.15%,0.57 %;表明该测定方法具有良好的重复性。再取上述供试品溶液1份,于0,4,8,12,24 h 分别进样,每次进样10 μL,测定供试品各成分的峰面积。结果,供试品中芍药苷、黄芩苷、二苯乙烯峰面积的RSD 分别为0.76 %,0.80%,0.59 %。说明益肾降糖胶囊供试品溶液中芍药苷、黄芩苷、二苯乙烯苷用该方法测定在24 h 内保持稳定。2.2.8
加样回收率试验 取上述重复性试验测定的益肾降糖胶囊6份,每份0.17 g,精密称定,分别置于10 mL 容量瓶中,精密加入芍药苷对照品贮备液2 mL、二苯乙烯苷对照品贮备液0.15 mL,黄芩苷对照品贮备液2.3 mL,按照供试品溶液制备方法制备加样回收率试验供试品溶液,按“2.2.1”项下设定的色谱参数进样测定供试品中芍药苷、二苯乙烯苷、黄芩苷的含量,并计算各成分的加样回收率。芍药苷、黄芩苷、二苯乙烯苷、的平均回收率分别为98.96 %,99.91 %,99.89%;RSD 分别为0.76 %,1.16%,1.41%;表明采用该方法测定益肾降糖胶囊中芍药苷、黄芩苷、二苯乙烯苷的含量,回收率良好,结果准确可靠。结果见表1。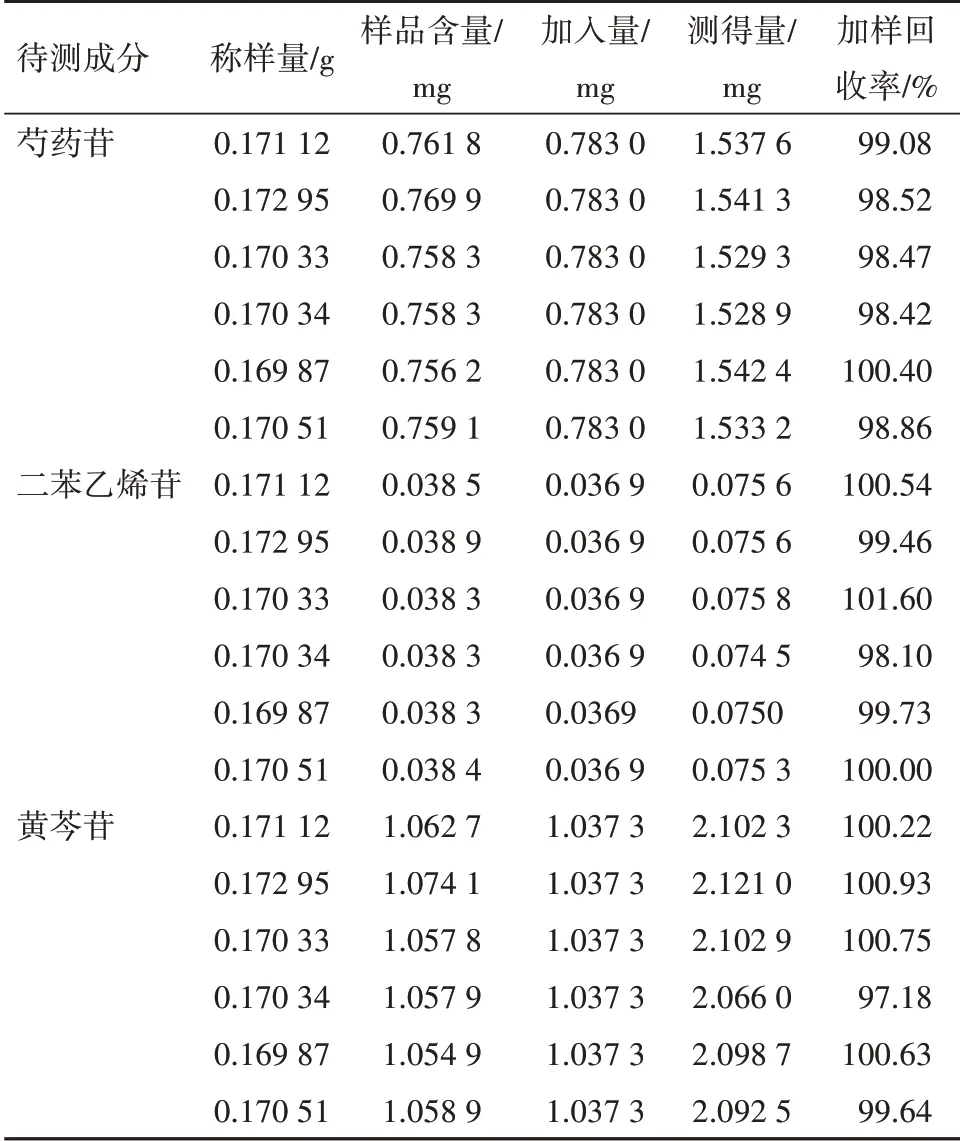
表1 加样回收率试验结果(n=6)
2.2.9
样品的测量 分别取不同批次的益肾降糖胶囊3 批,按照“2.2.3”的方法制备益肾降糖胶囊供试品溶液3 份,按“2.2.1”项下设定的色谱参数测定供试品溶液中芍药苷、二苯乙烯苷、黄芩苷的峰面积,并以“2.2.5”项下各成分的标准曲线计算含量,见表2。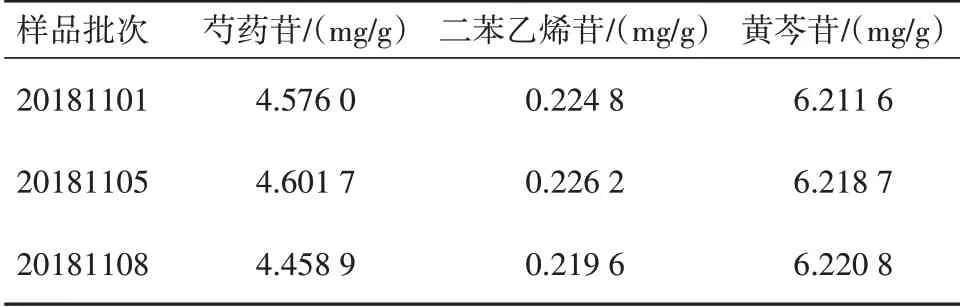
表2 样品含量测定结果(n=3)
3 讨论
3.1 薄层鉴别药味的选择
预试验中,对方中的10味药材进行了薄层色谱鉴别,当归、苍术的对照药材有荧光斑点,而样品在相应位置无相同的荧光斑点;方中肉苁蓉的鉴别,供试品与对照药材在相同位置有相应的斑点,但斑点不清晰,拖尾明显;方中太子参的鉴别,按《中国药典》一部(2015年版)太子参项下的方法进行检测,结果薄层色谱显示,供试品与对照药材在相应位置上显同样颜色的斑点,但阴性对照样品有同样的干扰斑点;故当归、苍术、肉苁蓉、太子参未纳入现质控方法中。其他6 味药的薄层鉴别图谱斑点清晰可见,分离度良好,专属性强;其中赤芍与玄参是在原标准基础上,进行优化,同时检测,简化了步骤,节约成本。3.2 提取方法的选择
含量测定项目下供试品溶液的制备在提取时,提取溶剂分别考察了甲醇、50%甲醇、稀乙醇和70%乙醇4种溶剂;提取方法采用超声提取和水浴加热回流提取两种方法,提取时间考察了15,30,45和60 min,结果显示采用稀乙醇超声提取30 min,测得各成分含量最高,且该提取方法操作简单可行,故最终提取方法采用稀乙醇超声提取30 min。3.3 检测波长的选择
芍药苷、二苯乙烯苷、黄芩苷的最大吸收波长分别为230,280,320 nm。230,280 nm处3个成分均有吸收,320 nm处芍药苷无吸收;其中二苯乙烯苷在230、280 nm 处的吸收较小,且样品中二苯乙烯苷含量较低,故选择其最大吸收波长320 nm 作为测定波长;黄芩苷在320 nm,280 nm 处的吸收均较大,且在两波长处测量结果基本一致;芍药苷在230 nm处吸收最强,故最终确定230 nm波长测定芍药苷,320 nm波长测定二苯乙烯苷和黄芩苷。3.4 流动相的选择
流动相的组成及比例对样品峰的峰型及出峰时间都有很大的影响,曾考察了甲醇-水、乙腈-水、甲醇-0.1%磷酸溶液、乙腈-0.1%磷酸溶液、甲醇-0.2%磷酸溶液、乙腈-0.2%磷酸溶液等体系,在试验过程中发现,采用甲醇-0.2%磷酸溶液体系时,三成分峰型比较对称,故选择作为流动相体系。多次试验发现甲醇-0.2%磷酸溶液的比例大于40∶60 时,芍药苷与二苯乙烯苷出峰时间接近,两成分无法完全分开,而流动相比例低于40∶60时,黄芩苷的出峰时间在60 min 左右,检测时间较长。综合考虑各因素,最终确定梯度洗脱,三种成分得到良好的分离,且检测时间控制在40 min 以内,提高了效率。综上所述,本质控方法可操作性强,具有良好的重现性,检测结果准确度高,可用于益肾降糖胶囊的质量控制。



