苟惠天,薛慧文,殷 宏,孙晓林*,罗建勋*
(1甘肃农业大学动物医学院,兰州 730070;2中国农业科学院兰州兽医研究所,兰州 730046)
基于COⅠ基因序列对我国部分巴贝斯虫分类的研究
苟惠天1,薛慧文1,殷宏2,孙晓林1*,罗建勋2*
(1甘肃农业大学动物医学院,兰州 730070;2中国农业科学院兰州兽医研究所,兰州 730046)
为了确定巴贝斯虫中国分离株的分类地位,对我国报道的7种巴贝斯虫11个地方分离株的COⅠ基因序列进行测定;并与GenBank中其他巴贝斯虫COⅠ和相应18S rRNA基因序列分别构建系统发生树,比较基于不同基因的分类结果。结果显示:11株巴贝斯虫的COⅠ基因大小在935~999 bp,与18S rRNA基因比较,COⅠ基因序列含有较多的变异位点及简约信息位点;基于两个基因的系统发育树,对于牛巴贝斯虫的分类结果基本一致。但也存在如下差异:18S rRNA无法将泰勒虫与巴贝斯虫进行区分,而COⅠ基因可明显区分;羊巴贝斯虫新疆未定种的分类地位在基于COⅠ基因的分类中更具合理性。来源于COⅠ和18S rRNA的信息都说明我国莫氏巴贝斯虫的不同地方株间可能存在亚种关系。该研究为巴贝斯虫的分子分类提供了候选基因。
巴贝斯虫;分类;系统发育树;COⅠ基因
巴贝斯虫(Babesiaspp.)属于顶复门、梨形虫纲、巴贝斯虫科、巴贝斯虫属,是一类能引起人和动物巴贝斯虫病的蜱传性血液原虫。该病呈全球分布,给公共健康和畜牧业发展带来重大影响[1]。在中国引起牛巴贝斯虫病的病原有双芽巴贝斯虫(Babesiabigemina)、牛巴贝斯虫(B.bovis)、卵形巴贝斯虫(B.ovata)、大巴贝斯虫(B.major)、东方巴贝斯虫(B.orientalis)和巴贝斯虫未定种喀什分离株(BabesiaU sp.Kashi)。已证实微小扇头蜱、长角血蜱、刻点血蜱、镰形扇头蜱、小亚璃眼蜱及其他一些硬蜱可以传播这些巴贝斯虫[2-3]。引起羊巴贝斯虫病的病原有绵羊巴贝斯虫(B.ovis)、莫氏巴贝斯虫(B.motasi)和巴贝斯虫未定种新疆分离株(Babesiasp.Xinjiang);已知青海血蜱、长角血蜱和小亚璃眼蜱是以上三种巴贝斯虫的传播媒介[4-5]。
近年来,18S rRNA作为分子标记在巴贝斯虫的研究中得到了广泛应用[6]。但是由于物种多样性以及单个标记基因很难真实反映物种间的进化关系。所以一些新的标记基因也被用于此类研究,诸如转录间隔区(ITS)及主要表面蛋白基因(MPSP)的应用[7-8]。细胞色素C氧化酶基因Ⅰ(COⅠ)作为线粒体基因组的重要组成部分,较之于核糖体基因,更能真实地反映物种的遗传信息,因此近年来被广泛地用于物种的系统进化分析及生物条形码研究中[9-10]。但迄今为止,有关以COⅠ作为标记基因对巴贝斯虫的进化分析的研究,在国内外还未见有报道。笔者试图通过比较基于两个分类基因COⅠ和18S rRNA在巴贝斯虫中的进化关系,进一步了解巴贝斯虫不同虫株之间的遗传进化关系[11]。
1 材料与方法
1.1虫株和实验动物
本试验涉及的11株巴贝斯虫均分离自中国。包括六株牛巴贝斯虫和五株羊巴贝斯虫。以上虫株在实验室液氮保存,详细资料见表1。
6~12月龄的黄牛和绵羊购自无巴贝斯虫病报道的地区。在试验前30 d所有的实验动物摘除脾。在试验前10 d,所有的动物取耳尖血制备涂片,再次检查是否有巴贝斯虫感染。只有检查结果为阴性的动物,方可用于后续试验。
1.2DNA提取、目的基因扩增及测序
对6只黄牛和5只绵羊分别感染含有不同巴贝斯虫分离株的虫血各10 mL。当染虫率大于5%时,静脉采血,分离红细胞。使用DNA提取试剂盒(QIAGEN,德国)进行巴贝斯虫基因组的提取,方法按说明书操作,分光光度计测算DNA的浓度。以未感染梨形虫的动物血液作为阴性对照。
利用GenBank已公布的巴贝斯虫COⅠ基因的序列,设计两对引物。BaF1(ORF第91位):5′-ATAGGATTCTATATGAGTAT-3′,BaR1(ORF第1 318位):5′-ATAATCAGGTATTCTCCTTGG-3′;BaF2(ORF第190位):5′-TCTCTTCATGGTTTAATTATGATAT-3′,BaR2(ORF第1 315位):5′-TA-GCTCCAATTGATAAAACAAAGTG-3′。PCR扩增按照50 μL体系进行,其中基因组DNA 100 ng,上下游引物各1 pmol·L-1,dNTP 160 μmol·L-1,TaqDNA聚合酶1 U,MgCl21.75 mmol·L-1,最后用灭菌水补足50 μL,以上试剂均购自大连宝生物公司。PCR反应条件:94 ℃预变性5 min;94 ℃变性1 min,63 ℃或58 ℃退火1 min,72 ℃延伸3 min,35个循环;72 ℃终延伸8 min,最后4 ℃保存。取5 μL PCR产物再进行电泳,观察结果。
切胶纯化PCR产物,然后将目的片段克隆到pGEM-T easy载体(Promega,美国)。为确保结果的准确性,每株巴贝斯虫送5个鉴定为阳性的克隆到测序公司,用ABI 3730进行序列测定。另外,对测序巴贝斯虫的相应18S rRNA基因序列,分别从GenBank中进行下载。
1.3序列比对和进化分析
使用Lasergene软件包中的SeqMan对每一条测序结果进行拼接,并将拼接结果在NCBI中作BLAST比对,检测是否混杂有其他DNA。同时以两株泰勒虫(Theileriaparva和Theileriasergenti)的COⅠ基因序列作为外群。使用MEGA4中的多序列比对方法,分别对COⅠ和18S rRNA两组序列进行比对,使用K2-P方法计算遗传距离[12]。用最大简约法(MP)和贝叶斯法(BI)分别针对COⅠ和18S rRNA序列构建系统发育树。在最大简约法中,使用TBR启发式搜索的方法对数据进行分析,空位被视为缺失,颠换和转换具有相等的权重,自展分析次数为1 000[13]。在贝叶斯法中,分析采用GTR+I+Γ模型,四条链分别进行计算,COⅠ和18S rRNA各运算2.0×105代后数据得到收敛[14]。最后将两种方法产生的系统发育树用Tree View软件显示,并进一步编辑。
2 结 果
2.1COⅠ基因的扩增
从11株巴贝斯虫基因组中均扩增到COⅠ基因序列,大小为935~999 bp,经Blast N比对无误后登录GenBank,序列号见表1。
表1本研究中所用到COⅠ 和18S rRNA的序列信息
Table 1Origins of the piroplasma and GenBank accession numbers for theCOⅠ and 18S rRNA gene used in this study

名称Species分离地Origin宿主Host序列Type长度/bpLength登录号GenBanknumber序列来源SequenceSourceB.major中国新疆中国新疆牛COⅠ18S9351693JQ518310AY603399本研究扩增GenBank中下载B.ovata中国河南中国河南牛COⅠ18S9761685JQ518306AY603401本研究扩增GenBank中下载B.ovata中国甘肃中国甘肃牛COⅠ18S9771687JQ518307AY603400本研究扩增GenBank中下载B.bigemina中国云南中国云南牛COⅠ18S9771693JQ518300AY603402本研究扩增GenBank中下载B.bigemina日本中国牛COⅠ18S14321693AB499085HQ840960GenBank中下载GenBank中下载B.bovis中国河南中国河南牛COⅠ18S9641653JQ518301AY603398本研究扩增GenBank中下载B.bovis日本美国牛COⅠ18S14341588AB499088HQ264112GenBank中下载GenBank中下载B.bovis美国美国牛COⅠ18S14341588EU075182HQ264111GenBank中下载GenBank中下载BabesiaUsp.中国新疆中国新疆牛COⅠ18S9771689JQ518308AY726556本研究扩增GenBank中下载B.motasi中国甘肃中国甘肃羊COⅠ18S9771683JQ518302AY260181本研究扩增GenBank中下载B.motasi中国甘肃中国甘肃羊COⅠ18S9771687JQ518303AY260182本研究扩增GenBank中下载B.motasi中国甘肃中国甘肃羊COⅠ18S9771680JQ518304DQ159072本研究扩增GenBank中下载B.motasi中国河北中国河北羊COⅠ18S9771683JQ518305DQ159074本研究扩增GenBank中下载Babesiasp.中国新疆中国新疆羊COⅠ18S9761715JN391437DQ159073本研究扩增GenBank中下载B.caballi南非南非马COⅠ18S14341694AB499086Z15104GenBank中下载GenBank中下载B.gibsoni日本日本狗COⅠ18S14341707AB499087EU084677GenBank中下载GenBank中下载T.parva日本日本水牛COⅠ18S14391583AB499089HQ895969GenBank中下载GenBank中下载
2.2COⅠ基因和18S rRNA基因的序列比对
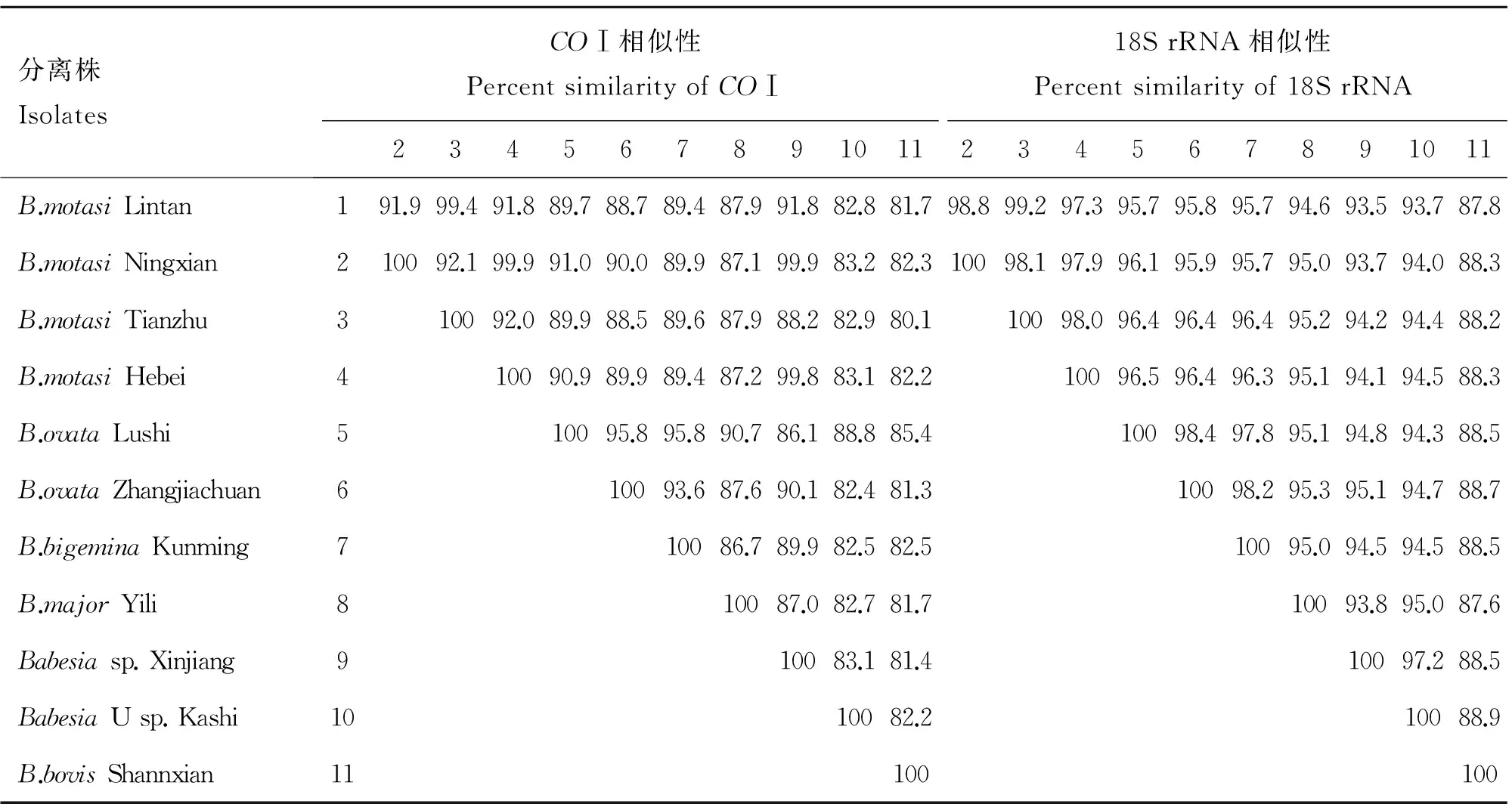
从GenBank中下载到18S rRNA基因的大小为1 583~1 715 bp。所有序列经Claustal W比对后,得出18S rRNA基因含有1 563个信息位点,其中变异位点308个,248个为简约信息位点;COⅠ基因中含有923个信息位点,其中变异位点354个,简约信息位点281个。基于两个基因不同种之间,同一种不同分离株之间的序列相似性见表2。
2.3系统进化分析
为了确定巴贝斯虫的系统发生关系,分别对COⅠ和18S rRNA基因序列用MP法和BI法构建了系统发育树。由于每一个基因其MP树和BI树的拓扑结构几乎一致,所以用合一树来表示。图1中A、B分别代表COⅠ和18S rRNA的合一树。通过比较,基于两个基因的系统发育树,都能真实反应牛巴贝斯虫间的分类地位。但也存在如下差异:发现18S无法将泰勒虫与巴贝斯虫进行很好区分,而COⅠ基因可明显区分;羊巴贝斯虫新疆未定种的分类地位在两个发育树中存在较大差异。
表2巴贝斯虫中国分离株COⅠ和18S rRNA基因序列的相似性比对
Table 2Sequence similarity ofCOⅠand 18S rRNA ofBabesiaspp.from China isolates
%
3 讨 论
国内外有关巴贝斯虫的分子分类学研究主要依靠18S rRNA基因。但近年来越来越多的研究显示,18S rRNA作为分类基因在科或更高阶元的研究时,显示出一定优势;在本研究中,18S rRNA变异位点主要集中在少数几个区域,如:第157—216位,第568—625位,第701—733位,第1 233—1 259位,其余部位则为高度保守。由于其序列的高度保守性,在区分亲缘关系很近的物种(属以下)时,其结果不甚理想。线粒体基因组,特别是COⅠ基因,因其独立的进化机制,在很大程度上保证了物种遗传信息的真实性。另外,具有适度的序列变异性,使其在任何分类阶元中都可使用。因此,国内外越来越多的学者将其作为新的分类基因。国际DNA条形码联盟更是推荐COⅠ为首选基因,进行物种的分子分类。与18S rRNA比较,COⅠ基因的变异位点比例更高,但整段序列并没有明显的保守区和变异区,而是变异位点和保守位点交替排列。因此,基于COⅠ基因的巴贝斯虫分类,能够更清楚地反映出巴贝斯虫与泰勒虫的分类地位,不同宿主巴贝斯虫之间、同一种巴贝斯虫不同分离株间的关系[15]。
有关羊的巴贝斯虫在之前的研究中主要有两种:莫氏巴贝斯虫(B.motasi)和羊巴贝斯虫(B.ovis),其传播媒介分别是刻点血蜱和囊性扇头蜱,随后的研究发现粗糙巴贝斯虫(B.crassa)也可感染羊。有关莫氏巴贝斯虫的分类,有学者认为其存在亚种。Uilenberg提出至少可以将欧洲的巴贝斯虫分为高致病性和低致病性两个类群[16-17]。笔者实验室在过去的研究中,从不同地区绵羊和山羊的血液样品中曾分离到几株大型巴贝斯虫[4,18]。这些巴贝斯虫后来被命名为莫氏巴贝斯虫临潭株(B.motasiLintan)、莫氏巴贝斯虫宁县株(B.motasiNingxian)、莫氏巴贝斯虫天祝株(B.motasiTianzhu)和莫氏巴贝斯虫河北株(B.motasiHebei)。通过COⅠ系统发育树,可以看到临潭株和天祝株落在一个分枝中,河北株和宁县株在另一分枝中。该结果与18S rRNA的进化树不太一致,但是与ITS分析的结果相一致。宁县株是从甘肃省东部地区的绵羊血液之中分离得到的,对绵羊和山羊具有高致病性,其形状与欧洲的莫氏巴贝斯虫非常相似,长角血蜱是已知该巴贝斯虫的传播媒介。临潭株可被青海血蜱和长角血蜱传播,并且具有低致病性。河北株和天祝株的生物学特性尚不十分清楚,但是它们都分离自血蜱属分布的地区[17]。此外,以临潭株体外培养物经纯化后作为抗原,进行的ELISA试验结果显示,与宁县株和河北株的阳性血清不发生交叉反应,但却可以与天祝株的阳性血清具有很强的交叉反应[19]。以上结果说明我国莫氏巴贝斯虫的不同地方株间很可能存在亚种关系。
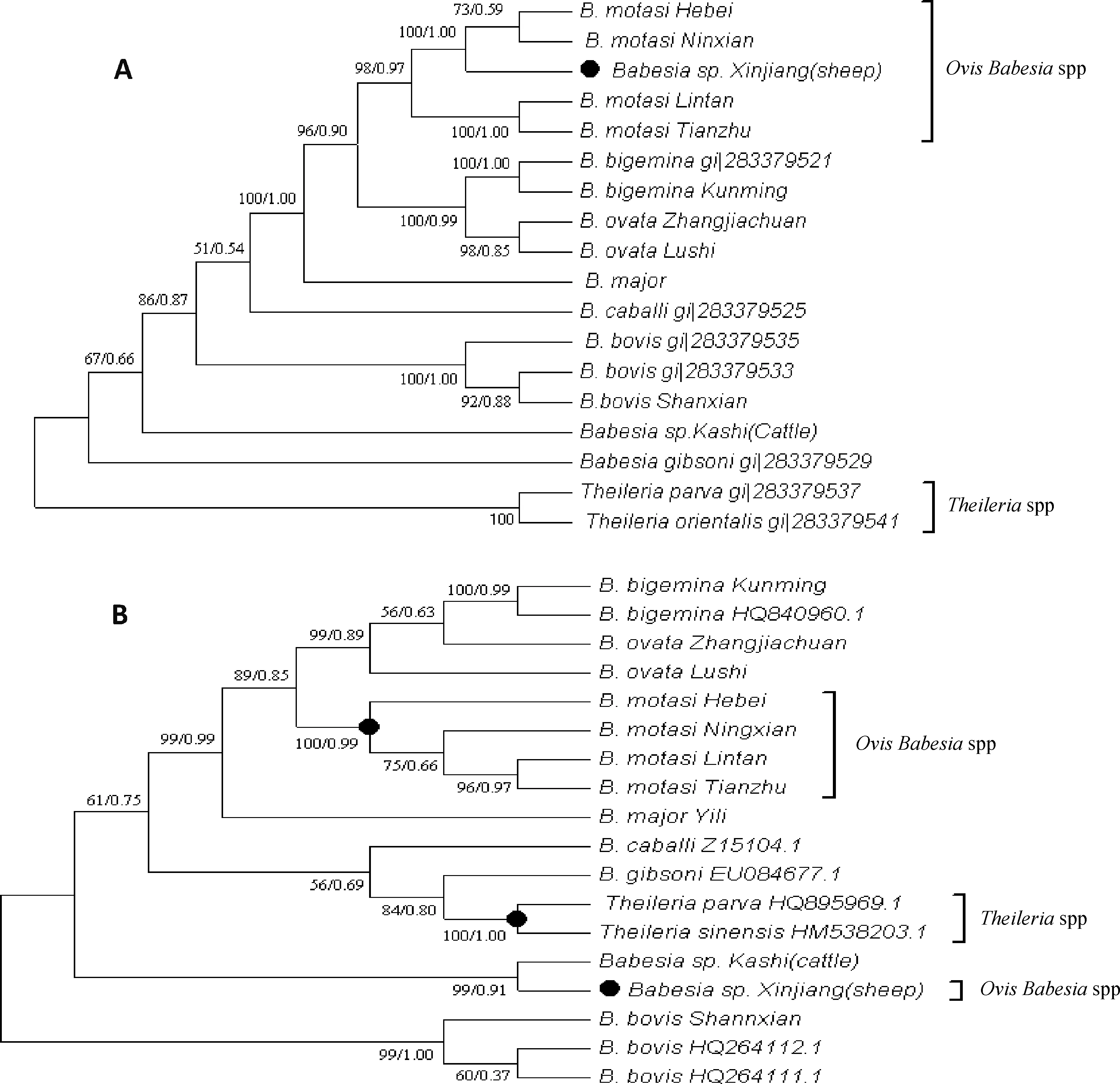
A.COⅠ基因序列;B.18S rRNA基因序列;系统发育树的构建采用MP和BI两种计算方法,节点数字分别表示自展值和后验概率;两株泰勒虫作为外群These trees were calculated using the BI and MP methods.Trees were constructed with COⅠ sequences (A) and 18S rRNA sequences (B).The bootstrap values supporting and posterior probability each node are shown.Two Theileria isolates were usedas outgroup图1 基于COⅠ和18S rRNA基因的巴贝斯虫系统发育树Fig.1 Phylogenetic trees of Babesia isolates based on their COⅠand 18S rRNA genes
巴贝斯虫新疆未定种(Babesiasp.Xinjiang)是笔者实验室于2001年从新疆采集的血红扇头蜱和小亚璃眼蜱,感染绵羊后,经由羊血中分离得到的一种巴贝斯虫。综合形态学观察,传播试验等生物学特性,确定该巴贝斯虫是独立于莫氏巴贝斯虫、绵羊巴贝斯虫、粗糙巴贝斯虫之外的一个新种[17]。本研究中,基于18S rRNA的系统发育树中,Babesiasp.Xinjiang与其他四株羊巴贝斯虫的位置较远,反而与一株同样分离自新疆,感染牛的巴贝斯虫(Babesiasp.Kashi)归为一支。该结果的出现可能与构建系统发育树所用的序列相关。而在基于COⅠ的系统发育树中,该未定种与莫氏巴贝斯虫归为一支,并位于致病性不同的四株莫氏巴贝斯虫之间,显示与莫氏巴贝斯虫存在很近的遗传关系。综合以上18S rRNA和COⅠ的结果,都未能真实反应该巴贝斯虫未定种的分类地位。因此,对于该未定种的命名还需更多生物学数据进行佐证[11]。
总之,与之前基于18S rRNA和其他基因的研究结果比较,COⅠ基因的应用,可以帮助更好地解释巴贝斯虫各种之间的进化关系。但由于COⅠ基因在巴贝斯虫上研究非常少,以致基因数据库中能用到的数据量还很有限。因此,通过本研究,希望能引起广大研究者的兴趣,在以后的分类研究中引入COⅠ基因,推动巴贝斯虫甚至梨形虫的分类研究。
[1]TELFORD S R 3rd,SPIELMAN A.Reservoir competence of white-footed mice forBabesiamicroti[J].JMedEntomol,1993,30(1):223-227.
[2]LUO J,YIN H,GUAN G,et al.A comparison of small-subunit ribosomal RNA gene sequence of bovineBabesiaspecies transmitted byHaemaphysalisspp. in China[J].ParasitolRes,2005,95(2):145-149.
[3]YIN H,LU W,LUO J,et al.Experiments on the transmission ofBabesiamajorandBabesiabigeminabyHaemaphysalispunctata[J].VetParasitol,1996,67(1-2):89-98.
[4]GUAN G Q,YIN H,LUO J X,et al.Transmission ofBabesiasp to sheep with field-collectedHaemaphysalisqinghaiensis[J].ParasitolRes,2002,88(13 Suppl 1):22-24.
[5]NIU Q,LUO J,GUAN G,et al.Differentiation of two ovineBabesiabased on the ribosomal DNA internal transcribed spacer (ITS) sequences[J].ExpParasitol,2009,121(1):64-68.
[6]AHMED J S,LUO J,SCHNITTGER L,et al.Phylogenetic position of small-ruminant infecting piroplasms[J].AnnNYAcadSci,2006,1081:498-504.
[7]LIU J,YIN H,LIU G,et al.Discrimination ofBabesiamajorandBabesiaovatabased on ITS1-5.8S-ITS2 region sequences of rRNA gene[J].ParasitolRes,2008,102(4):709-713.
[8]LIU A,GUAN G,LIU Z,et al.Detecting and differentiatingTheileriasergentiandTheileriasinensisin cattle and yaks by PCR based on major piroplasm surface protein (MPSP)[J].ExpParasitol,2010,126(4):476-481.
[9]MATZEN DA SILVA J,CREER S,DOS SANTOS A,et al.Systematic and evolutionary insights derived from mtDNA COI barcode diversity in the decapoda (Crustacea:Malacostraca)[J].PLoSOne,2011,6(5):e19449.
[10]DERYCKE S,VANAVERBEKE J,RIGAUX A,et al.Exploring the use of cytochrome oxidase c subunit 1 (COI) for DNA barcoding of free-living marine nematodes[J].PLoSOne,2010,5(10):e13716.
[11]ARMSTRONG P M,KATAVOLOS P,CAPORALE D A,et al.Diversity ofBabesiainfecting deer ticks (Ixodesdammini)[J].AmJTropMedHyg,1998,58(6):739-742.
[12]TAMURA K,DUDLEY J,NEI M,et al.MEGA4:molecular evolutionary genetics analysis (MEGA) software version 4.0[J].MolBiolEvol,2007,24(8):1596-1599.
[13]SWOFFORD D.PAUP 4.0 b10:Phylogenetic analysis using parsimony[M].Sunderland,MA,USA:Sinauer Associates,2002.
[14]RONQUIST F,HUELSENBECK J P.MrBayes 3:Bayesian phylogenetic inference under mixed models[J].Bioinformatics,2003,19(12):1572-1574.
[15]MEDINA M,COLLINS A G,SILBERMAN J D,et al.Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA[J].ProcNatlAcadSciUSA,2001,98(17):9707-9712.
[16]UILENBERG G.Babesia—a historical overview[J].VetParasitol,2006,138(1-2):3-10.
[17]BAI Q,LIU G,LIU D,et al.Isolation and preliminary characterization of a largeBabesiasp.from sheep and goats in the eastern part of Gansu Province,China[J].ParasitolRes,2002,88(13 Suppl 1):S16-S21.
[18]LIU A H,YIN H,GUAN G Q,et al.At least two genetically distinct largeBabesiaspecies infective to sheep and goats in China[J].VetParasitol,2007,147(3-4):246-251.
[19]GUAN G,MOREAU E,LIU J,et al.Babesiasp.BQ1 (Lintan):Molecular evidence of experimental transmission to sheep byHaemaphysalisqinghaiensisandHaemaphysalislongicornis[J].ParasitolInt,2010,59(2):265-267.
(编辑白永平)
Toxonomic Study of SomeBabesiaspp.Based onCOⅠGene in China
GOU Hui-tian1,XUE Hui-wen1,YIN Hong2,SUN Xiao-lin1*,LUO Jian-xun2*
(1.CollegeofVeterinaryMedicine,GansuAgriculturalUniversity,Lanzhou730070,China;2.LanzhouVeterinaryResearchInstituteofChineseAcademyofAgriculturalScience,Lanzhou730046,China)
In order to clarify the taxonomic status ofBabesiaspp,theCOⅠgene were sequenced from 11 isolates of 7 species reported in China;The phylogenetic tree were constructed usingCOⅠand 18S rRNA sequences downloaded from GenBank and this study,and also the two phylogenetic trees were compared.As result shown,the length ofCOⅠgene was 935-999 bp,and theCOⅠgene had more variation and informative sites than 18S rRNA.The taxonomic status of bovisBabesiaspp.based onCOⅠand 18S rRNA shared high similarity.However,there were also some differences from two phylogenetic trees.Such as,theCOⅠgene can differentiateBabesiaspp andTheileriaspp clearly.To 18S rRNA,it was more difficult.The taxonomic status based onCOⅠgene ofBabesiasp.from Xinjiang was more reasonable.The information of two phylogenetic tree suggested that theBabesiamotasiisolated from different region may represent different subspecies.This study supported the candidate gene of molecular taxonomy ofBabesiaspp.
Babesiaspp.;toxonomy;phylogenetic tree;COⅠgene
10.11843/j.issn.0366-6964.2016.06.028
2015-11-16
国家自然科学基金(31560700);甘肃省自然科学基金(145RJZA172);兰州市科技创新人才计划(2014-2-11);甘肃农业大学盛彤笙创新基金(GSAU-STS-1422)
苟惠天(1980-),甘肃临洮人,讲师,博士,主要从事兽医公共卫生研究工作,E-mail:gouht@gsau.edu.cn
罗建勋,男,研究员,E-mail:vectorparasit@126.com;孙晓林,男,教授,E-mail:sunxl@gsau.edu.cn
S852.72
A
0366-6964(2016)06-1293-06



