张跃博,颜 华,王立刚,赵福平,侯欣华,刘 欣,高红梅,张龙超,王立贤
(中国农业科学院北京畜牧兽医研究所,北京 100193)
传统观念认为RNA仅忠实转录DNA,但随着试验技术的不断进步和研究的持续深入,一系列复杂的转录后加工修饰过程逐渐被证实。生物体通过对这些过程的调控来维持其体内转录本的稳定,并对相应的细胞内外刺激做出响应[1-2]。目前,已发现的RNA修饰方式有160多种[3]。
RNA编辑最早发现于1986年,Benne等[4]在锥体虫线粒体中发现细胞色素基因(CoxⅡ)的成熟mRNA上存在4个非线粒体基因组编码的碱基U,可抵消3′与5′编码区的读码框不融合现象,从而恢复CoxⅡ基因的原有功能,顺利翻译出具有活性的蛋白。起初,科学家认为此现象仅存在于这些稀有的原生动物中,但仅1年后,Powell等[5]和Chen等[6]发现,在哺乳动物载脂蛋白ApoB的转录产物中存在一个碱基U替换了原来的碱基C的现象。现在已知RNA编辑广泛分布于哺乳动物、原生动物、昆虫、植物、真菌、病毒等生物。mRNA测序计划表明,RNA编辑位点几乎存在于所有的转录本中,并可进行动态调控[7-8]。对RNA编辑检测和了解将有利于深入揭示生命过程的分子机理。截止到2018年1月22日,中国知网数据库中有关RNA编辑的综述仅33篇,主要介绍了RNA编辑的研究进展,涉及检测方法的内容极少。
本文将围绕RNA编辑的定义、功能以及RNA编辑位点检测方法等进行论述,并着重介绍目前广泛使用及最新开发的RNA编辑位点检测工具。
1 RNA编辑的定义
1986年,Benne等[4]发现,锥体虫线粒体CoxⅡ基因的mRNA上存在4个碱基插入现象,从而引入了RNA编辑这一概念。如今,RNA编辑被归纳为初级转录本上碱基的插入、缺失或者替换,是转录后发生的一种重要修饰,可改变RNA所携带的遗传信息,导致成熟RNA序列不同于其模板DNA[9]。RNA编辑增加了转录本及蛋白质的多样性,有利于生物体更好地适应生存环境,同时也对中心法则进行了补充和扩展,增进了人们对生物遗传规律的认知。
根据性质不同,RNA编辑可以划分为两类:碱基替换和碱基的插入或缺失。碱基插入或缺失型RNA编辑常见于单细胞真核生物,目前,哺乳动物中尚未见相关报道。哺乳动物中主要存在两种碱基替换型RNA编辑事件——A-to-I和C-to-U,前者在数量上具有绝对优势。A-to-I编辑是指腺苷A在作用于双链RNA的腺苷脱氨酶(adenosine deaminases acting on RNA, ADARs)的催化下,C6位氨基水解脱氨形成次黄苷I,而次黄苷在逆转录和翻译过程中被识别为鸟苷G,所以此过程又被称为A-to-G编辑(图1)[10]。C-to-U是指在胞苷脱氨酶家族APOBECs的作用下,胞苷水解脱氨形成尿苷,主要发生于3′UTR[11-13]。ApoB mRNA编辑是哺乳动物中发现的首个C-to-U编辑,其编辑机制被解析得也相对清楚,详细描述请见参考文献[14]。其它碱基替换型RNA编辑事件如U-to-C、G-to-A也均有报道[15],但它们是否真实存在还具有争议。哺乳动物中,RNA编辑主要具有如下特征:广泛存在于转录组,内含子和基因间隔区最为丰富[16];具有碱基偏好性,A-to-I编辑的5′端倾向于尿苷,3′端倾向于鸟苷[17];可成簇发生[18];具有时空特性,编辑水平具有随个体发育而升高的趋势[19];A-to-I编辑位点最多,占全部编辑位点的50%以上[15];位点特异性A-to-I编辑主要位于编码区或者保守区域[20];多发生于重复序列,在灵长动物中主要集中于Alu序列[21]。
2 RNA编辑酶
目前,哺乳动物中已知的RNA编辑酶主要有两类,即ADARs和APOBECs。ADARs起源于作用于tRNA的腺苷脱氨酶(adenosine deaminases acting on tRNA,ADAT),在脊椎动物中高度保守。APOBEC1是首个被克隆出的胞苷脱氨酶,与多个辅因子结合形成复合体催化胞苷脱氨反应。
2.1 ADARs
哺乳动物中主要存在3种ADAR蛋白,即ADAR1、ADAR2和ADAR3。三者均具有2~3个双链RNA结合功能域和1个腺苷脱氨催化功能域,但仅ADAR1和ADAR2具有催化活性。通过对具有催化活性的ADARs靶标偏好性研究发现,不同蛋白的RNA靶标存在明显差异[22],如ADAR1 主要催化重复区域位点,ADAR2主要编辑非重复编码区位点[23]。ADAR1具有两种亚型:ADAR1 p150和ADAR1 p110;两者均于生物体的各组织中广泛表达。ADAR1 p150由干扰素诱导表达,存在于细胞核和细胞质中;ADAR1 p110呈现连续表达,只存在于细胞核中[24]。敲除ADAR1基因的小鼠会因造血障碍和细胞的非正常凋亡而导致胚胎早期致死[25-26]。ADAR2主要位于细胞核中,以大脑中的分布最为丰富[27],其催化的腺苷脱氨反应通常具有位点特异性,如谷氨酸AMPA受体亚基GluR2上导致谷氨酰胺转变为精氨酸的A-to-I编辑[28-29],且可以通过编辑其转录本实现对自身活性的负调控[30]。
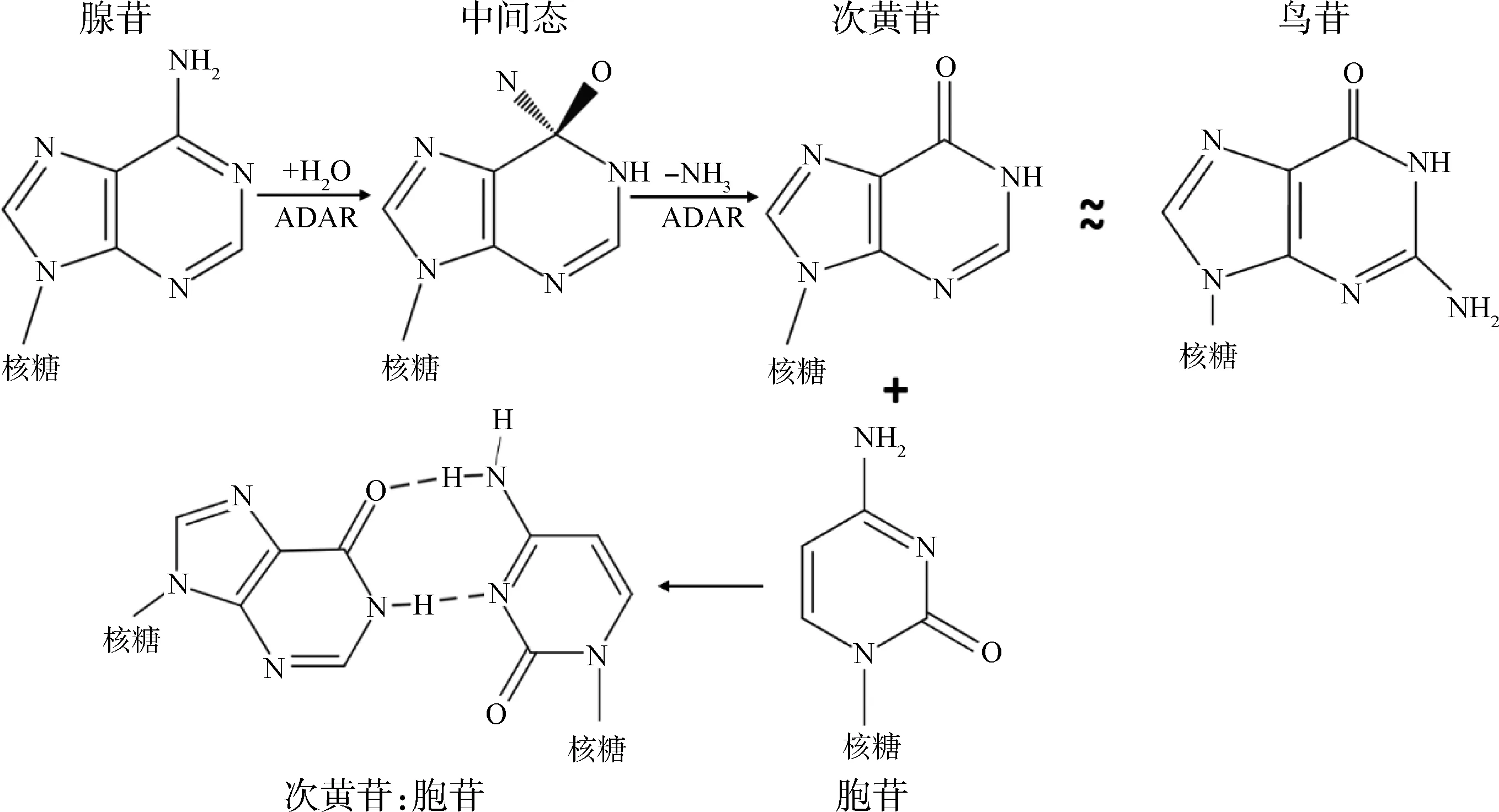
图1 A-to-I编辑的机制
Fig.1 The mechanism of A-to-I RNA editing
敲除ADAR2基因的小鼠在出生3周内会因癫痫发作而死[31]。ADAR3仅存在于大脑中,尚未有研究证明ADAR3直接参与RNA编辑,但其可以通过竞争性结合dsRNA抑制ADARs家族其他成员催化活性[32],具体作用机制有待进一步研究。
2.2 APOBECs
人的APOBECs家族具有11个成员,目前仅检测到APOBEC1、APOBEC3A和APOBEC3G具有催化C-to-U RNA编辑的脱氨酶活性[33-35]。APOBEC1定位于人1号染色体上,在小肠和肝中表达量最高,该基因敲除后小鼠的脂质代谢水平降低[36-37]。APOBEC3亚家族为哺乳动物特有,鼠只有一种APOBEC3基因,而猪、牛、羊、马和灵长类动物具有多种[38]。人的APOBEC3基因都位于22号染色体上,具有多种脱氨酶活性,在人进化过程中发挥重要作用[39-40]。APOBEC3A主要在骨髓细胞中表达[41]。APOBEC3G在多种组织中均表达[42],且具有2个脱氨酶功能域,而APOBEC1和APOBEC3A仅有1个脱氨酶功能域[34]。研究表明,APOBEC3A和APOBEC3G倾向作用于具有茎环结构(stem-loop structures)的RNA[43]。不同于ADARs,APOBECs需要与特定的辅因子结合才具有脱氨酶活性,APOBEC1的辅因子为RMB-47,而APOBEC3A和APOBEC3G的辅因子尚不清楚[44]。
尽管RNA编辑酶在RNA编辑中扮演至关重要的角色,但ADARs和APOBECs的表达量无法完美诠释编辑水平变异[45-47]。研究发现,编辑底物的序列特征在RNA编辑的发生及RNA编辑水平的调控中也发挥重要作用[41, 48]。因此,RNA编辑酶与RNA编辑位点周围的序列特征共同调控RNA编辑。
3 RNA编辑的功能
3.1 位于编码区的RNA编辑
RNA编辑广泛存在于转录组中,但发生在基因编码区的很少。编码区的RNA编辑事件可以导致蛋白重编码,产生非基因组编码的蛋白,在蛋白多样性的形成中发挥重要作用。目前,已明确生物学意义的RNA编辑位点多位于基因编码区。GluR-B上的Q/R位点在正常生理状态下几乎完全发生RNA编辑,使得原编码的谷氨酰胺转变为精氨酸,导致AMAP受体钙离子通透性显着下降,从而实现对细胞的钙平衡调节[49]。该Q/R位点的异常编辑可导致小鼠癫痫发作及死亡[31]。5-羟色胺受体基因HTR2C上的5个A-to-I编辑位点会影响HTR2C亚型的转运及其与G蛋白的偶联互作[50]。Kv1.1 I/V位点的编辑则明显降低该电压门控通道的失活率[51]。随着研究的深入,人们发现编码区的RNA编辑并非仅局限于神经受体和离子通道。例如,NEIL1上的编辑导致精氨酸转变成赖氨酸,影响了DNA修复酶NEIL1的特异性[52]。又如,GLI1第2179位核苷酸的编辑改变了该基因的转录效率,进而影响细胞增殖[53]。
3.2 位于非编码区的RNA编辑
近年来,诸多非编码区RNA编辑在生命调控中发挥的作用也逐渐得到揭示。A-to-I RNA编辑发生在转录后剪接前,而次黄苷I在剪接过程中被识别为鸟苷酸G,因此,A-to-I编辑可以创建新的剪接供体和受体,导致外显子滞留。ADAR2 mRNA前体内含子上的A-to-I编辑使得AA转换成AI,导致47 bp内含子序列滞留[30]。位于UTRs中的RNA编辑可以改变RNA的稳定性、亚细胞定位、翻译效率等[23]。发生在剪接增强子或沉默子等剪接调控元件上的RNA编辑可以影响剪接效率[54]。RNA编辑还可以调节miRNA的生成和靶向结合,如miRNA let-7g前体和miR-376a上的RNA编辑。miRNA let-7g的前体pri-let-7g上存在一个A-to-I编辑位点,敲除ADARB1后,鼠体内成熟的let-7g减少,导致其靶标基因Cry2的过表达,间接引起此老鼠的运动节律缩短[55]。miR-376a存在2个编辑位点,分别位于第4和44位碱基,只有第4位 碱基被编辑后,miR-376a才能靶向结合于PRPS1[56]。发生在lncRNA上的RNA编辑可以改变lncRNA的二级结构,并影响miRNA与lncRNA间的互作[57]。Yang等[58]研究发现,RNA编辑可在piRNA的生成中发挥调控作用。RNA编辑还可以修复基因功能,使在突变过程中出现遗传信息丢失的基因通过RNA编辑得以恢复。
此外,A-to-I编辑引入的肌苷还可以与肌苷特异性结合蛋白作用,发挥不同功能特性。例如,人核酸内切酶V能够特异性地作用于含有肌苷的RNA,促进被编辑转录本的降解[59]。
4 RNA编辑位点的检测方法
理论上,RNA编辑位点的检测并不复杂,通过比较RNA序列及其模板DNA序列,寻找二者间的差异位点,进而确定编辑位点。RNA编辑位点的检测依赖于测序技术,在刚出现DNA测序技术时,RNA编辑位点只是偶然得之,其鉴定工作进展缓慢。随着测序技术的进步以及测序结果的积累,越来越多的RNA编辑位点得以揭示,尤其是高通量测序技术的诞生,使得RNA编辑位点检出数量有了质的飞跃。RNA编辑具有多种检测方法,但延伸终止法、比较基因组法、基于EST序列的基因组序列比对法等均由于自身的局限性,现已很少使用。本文将着重介绍目前在RNA编辑研究中较常使用的几种检测方法。
4.1 Sanger测序图谱法
利用测序图谱来鉴别RNA编辑位点是最简便的方法,最早报道的RNA编辑现象由Benne等[4]利用Sanger测序研究锥体虫线粒体中的细胞色素基因时发现的。利用该方法识别RNA编辑时,先将RNA反转录为cDNA,再对待检测RNA区域cDNA及DNA进行PCR扩增并测序,如果DNA测序图谱中某位点为单一峰而在cDNA测序图谱中却是混合双峰,那么此位点就是一个RNA编辑位点[60]。根据杂合双峰的高度比可以直接估计该位点的编辑水平,与通过大量克隆测序获得的编辑水平基本相等[61]。目前,Sanger测序法仍广泛用于验证利用高通量测序鉴定出的RNA编辑位点[17, 23, 62-63]。尽管Sanger测序法准确性高,但通量低,难以实现在转录组水平上进行RNA编辑位点的检测,更适合于单序列的测序。
4.2 限制性内切酶酶切法
利用限制性内切酶的酶切反应也可以鉴定RNA编辑位点。该方法虽不宜鉴定新的RNA编辑位点,但对于已知的并且能够在编辑后产生新的或者是破坏原有酶切位点的RNA编辑位点的检测十分方便。例如,小鼠Serinc1基因chr10:57235791位点和Lars2基因chr9:123370996位点的编辑分别产生了限制性内切酶BspDI和RsaI的酶切位点[64]。该方法主要包括反转录、PCR扩增、酶切及凝胶电泳4步,其原理是限制性内切酶只能特异性地切开编辑后的PCR产物或者编辑前的PCR产物。利用灰度分析软件对电泳条带的宽窄强弱进行定量分析,还可得到特定编辑位点上编辑与未编辑产物的比例,进而计算出编辑水平[65]。该方法具有操作简单、易实现、耗时少、成本低的优点,适用于评估RNA编辑检测工具的可靠性,但不能用于检测未知RNA编辑位点。
4.3 化学试剂处理法
A-to-I编辑的特异之处在于腺苷A被编辑后形成次黄苷I,而I并不是RNA中常见的核苷。因此,可利用这一特性对RNA进行特定处理,从而检测I。自然状态下,RNase T1无法区分出I和G。1997年,Morse和Bass[66]先用乙二醛处理RNA,然后在高浓度硼酸环境下实现了RNase T1对I的特异性剪切。乙二醛与鸟苷G的N1和N2反应生成加合物,致使RNase T1无法识别,而高浓度硼酸使加合物更加稳定,但次黄苷I无法与乙二醛反应形成稳定的加合物。因此,经乙二醛处理后,RNase T1可对RNA上的I进行特异性剪切。Morse等[66-67]利用该方法在线虫中先后共发现了10个新编辑位点,在人脑中检测到了19个新编辑位点,并发现大多数位点位于重复元件中。2010年,Sakurai等[68]提出了另一种检测I的化学方法——肌苷化学消除法(inosine chemical erasing,ICE)。该方法使用丙烯腈处理RNA,使次黄苷氰通过迈克尔加成反应氰乙基化,导致次黄苷无法与胞苷配对,从而阻止次黄苷处的反转录,然后对处理和未处理的cDNA进行扩增测序,通过比较测序图谱,即可鉴定出RNA编辑位点。
4.4 高通量测序法
高通量测序技术的出现和发展,为研究RNA编辑事件提供了新的技术平台,其价格的大幅下降也为大规模研究RNA编辑事件提供了可能。自2009年起,人们开始利用新一代高通量测序技术在全转录组范围内鉴定RNA编辑位点[69]。目前,根据使用的测序数据不同,利用高通量测序技术研究RNA编辑事件的方法可以分成两种:一种是对来自于同一样品的全基因组(DNA-seq)和全转录组(RNA-seq)同时测序,通过比较两者间的差异,找到RNA-DNA差异位点;一种只需样本的转录本数据就可以识别该样本上的编辑位点。前者可以很好地区分发生在转录组水平上的RNA编辑位点和发生在基因组水平上的SNP,特别是已有数据库中未注释的SNP位点,但是需要对同一样品基因组和转录组同时测序,成本较高;后者的优点是成本相对较低,并且随着越来越多的测序数据被公开,科研工作者可以利用公共数据开展大规模多层次编辑事件研究,但存在难以彻底排除SNP干扰的缺点,鉴定结果假阳性率较高。
5 基于高通量测序的RNA编辑检测工具
基于高通量测序识别RNA编辑事件的方法流程复杂,需综合考虑多种影响因素对分析结果进行层层过滤,这对科研人员提出更高要求的同时也对非生物信息学专业科研从业者形成了技术壁垒。目前,已有多个专业分析工具被研发(表1),为科研人员进行RNA编辑研究提供了极大便利。
5.1 REDItools
REDItools主要包括REDItoolDnaRna.py、REDItoolDenovo.py和REDItoolKnown.py 3个脚本,均以预先比对好的BAM格式文件为输入文件[70]。REDItoolDnaRna.py要求同时具有RNA和DNA测序数据,通过检测二者序列间的差异鉴定RNA编辑位点,并通过推测位点所在链进而消除反义转录本及比对错误的影响,提高检测可靠性。REDItoolDenovo.py仅利用RNA测序数据检测编辑位点,并会根据碱基替换的经验分布对检测出的编辑位点进行Fisher精确检验。REDItoolKnown.py适用于检测已知RNA编辑位点在给定RNA测序数据中的编辑情况。该软件包提供了适用于不同情况下检测RNA编辑位点的工具及一系列用于位点过滤、注释等后续分析的工具,极大地方便了使用者,但在实际使用中运行较慢,尤其是在调用REDItoolBlatCorrection.py对比对结果进行校正时。
5.2 GIREMI
GIREMI(genome-independent identification of RNA editing by mutual information)是2015年发布的一款以BAM格式文件作为输入文件,只利用RNA测序数据进行RNA编辑检测的工具[71-72]。在RNA测序数据中,很多读长(reads)包含多个单核苷酸变异(single-nucleotide variants,SNVs),它们可能是基因组SNPs、RNA编辑位点或者测序比对错误。同一RNA测序读长或者读长对上的一对SNPs的单体型应与参考基因组DNA一致,但SNP和RNA编辑位点间则会随机组合。GIREMI通过计算SNVs间互信息(mutual information,MI)获得等位基因连锁水平,进而区分RNA编辑位点与SNPs,并利用广义线性模型提高检测效力。由于MI模型是专门针对二倍体生物构建且需要使用者提供已知SNPs数据以计算MI参考分布[73],GIREMI只适用于具有SNPs数据库的二倍体生物。
5.3 RED
RED(RNA editing sites detector)是第一款具有图形用户界面的RNA编辑位点检测工具[74]。该软件使用MySQL关系型数据库管理系统存储和查询大量数据,以提升软件性能。在DNA测序数据存在与否的情况下,均可使用RED对RNA测序数据进行分析并检测RNA编辑位点。该软件以BAM和VCF格式文件为输入文件,其中,BAM文件用于位点可视化,VCF文件用于检测RNA编辑位点,并且在无BAM文件情况下也可使用。此外,RED还需要使用者提供重复序列文件、gtf格式的注释文件、包含所有已知SNPs的VCF文件以及已知RNA编辑位点文件,用于数据过滤。RED灵敏度较低,与REDItools相差10%以上。
5.4 RES-Scanner
RES-Scanner将读长比对和位点检测整合,是一款简单高效的RNA编辑检测软件包[75]。使用者只需输入RNA和DNA测序产生的fastq原始文件,RES-Scanner便可自动调用BWA进行比对,并展开后续RNA编辑位点检测。该工具的最大特点在于引入了统计模型来推断DNA位点的基因型,而之前的工具或者分析流程均通过设定等位基因频率阈值判定DNA位点的基因型。对于初步获得的候选编辑位点,RES-Scanner通过调用BLAT对覆盖编辑位点的错配读长进行重新比对,以剔除因比对错误产生的假阳性位点,随后进行二项分布检验,并给出校正后的P值,以进一步降低因测序错误造成的假阳性。目前,RES-Scanner仅支持采用dUTP方法构建的链特异性文库,并且只能用于DNA和RNA测序数据同时存在的情况。
5.5 RNAEditor
RNAEditor是一款简单易用的RNA编辑位点检测软件,其既有命令行操作方式也有图形界面,并创新性地集成了能够检测编辑岛的聚类算法[76]。编辑岛(editing islands)即编辑位点富集区域,与单一编辑位点相比,其包含的RNA编辑位点可信度更高,且具有生物学意义的可能性更大。RNAEditor分析流程主要包括3部分,即比对、过滤和注释,其分析过程中依次调用BWA和GATK,分别用于读长比对和变异位点检测。虽然RNAEditor操作简单,用户只需提供RNA测序数据及参考基因组等必要文件,后续分析便可自动完成,但其不适用于无SNP数据的物种。
5.6 JACUSA
JACUSA(the JAVA framework for accurate SNV assessment)是通过比较DNA-RNA或RNA-RNA从而快速准确地鉴定RNA编辑位点的一款软件,可充分利用生物学重复测序数据[77]。该工具提供了参数M,用于设定变异位点上最大等位基因数。JACUSA于2017年更新到v1.2.0版本,开始支持链特异性双端测序数据。与REDItools相比,JACUSA虽然具有更高的可重复性,但也无法提供一站式服务,需要使用者预先完成读长比对。
5.7 RED-ML
RED-ML(RNA editing detection based on machine learning)是一款使用机器学习算法的能够充分利用各种来源信息优化过滤参数的RNA编辑位点检测工具[78]。该软件使用的信息主要分为3类:1)读 长信息,包括候选编辑位点的编辑水平和支持候选编辑位点的读长数;2)与测序错误和比对错误相关的信息,如碱基比对质量、候选编辑位点在读长中的位置、候选编辑位点是否位于简单重复序列等;3)已知的RNA编辑特征,如编辑类型、候选位点是否位于Alu区域及上下游碱基的倾向性等。RED-ML以BAM格式文件为输入文件,可以仅使用RNA测序数据检测RNA编辑位点,也可以结合DNA测序数据使用。因ML模型是使用在人淋巴母细胞中鉴定出RNA编辑位点及人RNA编辑所特有的各种特征训练而成[78],所以RED-ML目前只能用于检测人RNA编辑位点。此外,RED-ML无法检测编辑水平低于0.1的位点。
5.8 SPRINT
SPRINT(SNP-free toolkit for identifying RNA editing sites)是一款通过聚类单核苷酸变异对(SNV duplets)进行RNA编辑位点检测的工具[79]。单核苷酸变异对是指2个相邻的具有相同变异的单核苷酸变异位点。SPRINT利用编辑位点单核苷酸变异对与SNP单核苷酸变异对间的差异分布来区分RNA编辑位点和SNPs,从而摆脱了对已知SNPs的依赖。对于未成功比对的读长,SPRINT会将其与参考基因组上的A全部替换为G后重新比对,从而鉴定超RNA编辑位点(hyper RNA editing sites)。由于SPRINT是专门针对RNA测序数据设计的,DNA测序数据即使存在也无法使用。
此外还有一些网页版工具iRNA-AI[80]、PAI[81]、RCARE[82]、RASER[83]以及ExpEdit[84]等。Diroma等[73]利用模拟数据结合多种比对软件对REDItools、GIREMI、RES-Scanner、JACUSA和RNAEditor进行测评,发现REDItools和JACUSA软件在Alu区域检测到的编辑位点最多,而JACUSA和RES-Scanner软件对非Alu区的编辑位点的检测效果更好。
6 问题及展望
高通量测序技术的诞生帮助人们鉴定出大量的RNA编辑位点,也加深了人们对RNA编辑产生机理、调控机制及分布特征等的认识,但利用该方法鉴定RNA编辑位点仍存在一些技术问题:1)文库构建中的PCR扩增及测序过程存在一定错误率,导致鉴定出的RNA编辑位点假阳性升高[85];2)二代测序读长较短,加之重复序列的普遍存在及RNA的可变剪接等,导致读长定位不准确[86],产生假阳性RNA编辑事件;3)测序深度和均一性、建库方法、测序方法等都会影响结果准确性,非链特异性测序难以准确判定RNA编辑类型;4)参考基因组中仍存在许多gaps和错误,可导致读长回贴时出错;5)RNA 编辑水平通常较低,一般不超过20%,测序深度不足将会遗漏大量编辑事件;6)不同的工具或者同一工具的不同参数可能给出大相径庭的结果,导致研究人员无从选择。
表1RNA编辑位点检测工具的主要特征
Table1MainfeaturesofRNAeditingdetectiontools
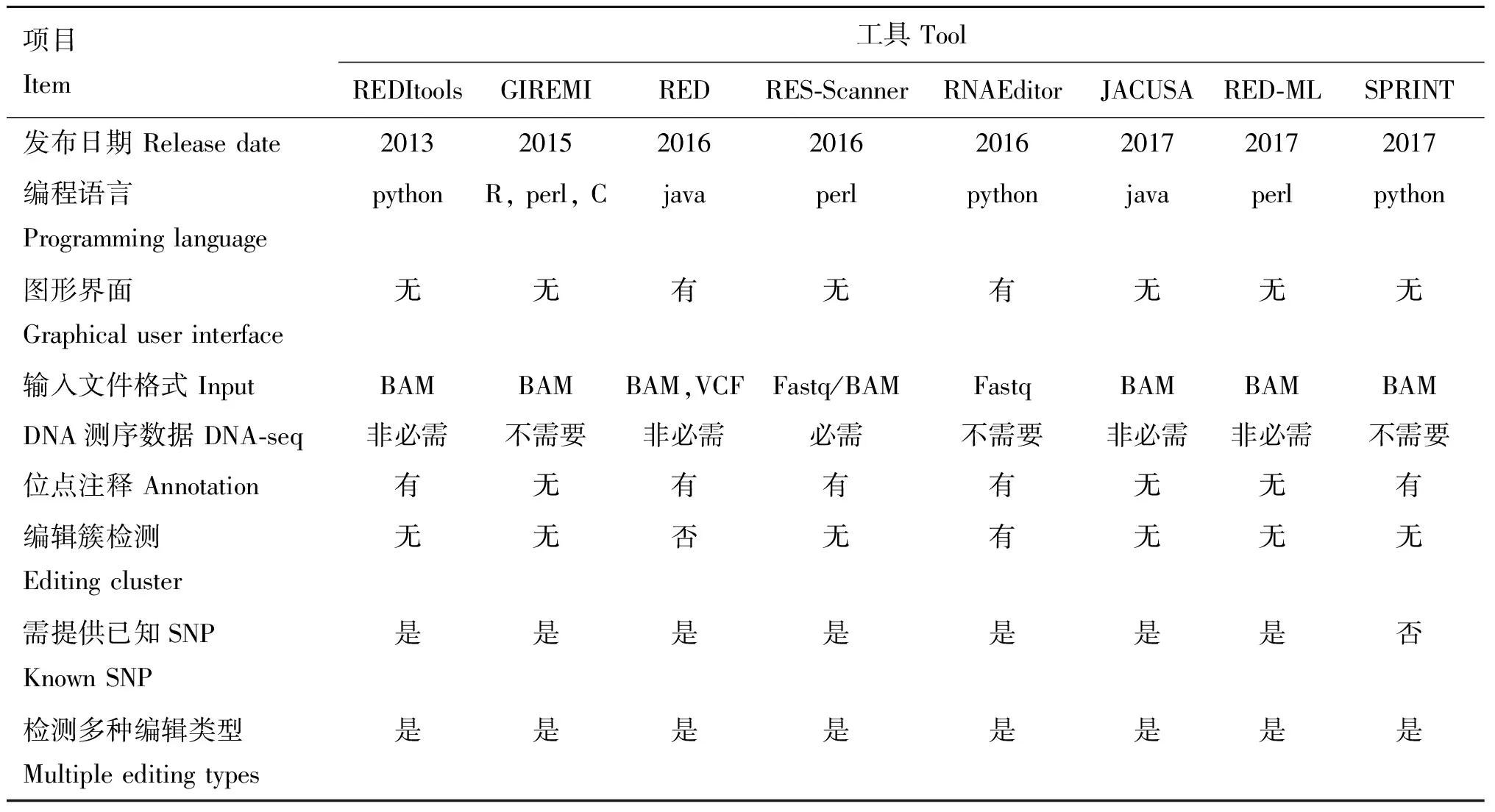
项目Item工具 ToolREDItoolsGIREMIREDRES-ScannerRNAEditorJACUSARED-MLSPRINT发布日期 Release date20132015201620162016201720172017编程语言Programming languagepythonR, perl, Cjavaperlpythonjavaperlpython图形界面Graphical user interface无无有无有无无无输入文件格式 InputBAMBAMBAM,VCFFastq/BAMFastqBAMBAMBAMDNA测序数据 DNA-seq非必需不需要非必需必需不需要非必需非必需不需要位点注释 Annotation有无有有有无无有编辑簇检测Editing cluster无无否无有无无无需提供已知SNPKnown SNP是是是是是是是否检测多种编辑类型Multiple editing types是是是是是是是是
为了解决这些问题,研究人员从不同角度出发综合考虑各种因素,设计了更加严格的分析流程:在试验设计中,采用具有更长读长和链特异性的双末端深度测序技术,设置生物学和技术重复;在读长比对时,选用可识别剪接的比对软件或同时将参考基因组和转录组作为回贴模板,提高回贴率;在RNA编辑事件鉴定时,采用多重过滤条件及复杂统计算法确保RNA编辑位点鉴定的特异性和灵敏度[73]。此外,能够提高鉴定准确性的策略也在不断涌现。在缺少基因组数据的背景下,研究人员利用RNA编辑的保守性、稀有SNP的低频性[87]和SNP间的连锁关系[71]鉴别RNA编辑事件与SNP。为克服高通量测序及后续分析中的弊端,科研人员开发了一些转录组预处理方法:利用肌苷特殊性,分别将肌苷特异性剪切和肌苷消除法与高通量测序结合,用以检测A-to-I RNA编辑位点,从而进一步排除因比对错误、测序错误、PCR错误等形成的G对鉴定造成的干扰;Zhang等[88]将显微射流复合PCR与深度测序结合,用于检测已知位点的编辑事件,克服了RNA-seq难以准确定量中低表达水平基因编辑的难题。三代测序技术的发展为更加准确鉴定重复区域内的RNA编辑事件带来了曙光,但目前三代测序错误率比较高[89],暂不适用于单碱基变异的检测。迅猛发展的生物信息学为人们提供了诸多简单易用的检测工具,但到底使用哪一款,还需要研究人员根据自身经费预算、试验设计和试验条件进行选择。
改革开放以来,我国畜禽育种取得了重大进展。在数量遗传学及群体遗传学理论指导下,综合应用分子生物学技术,多个符合国人消费需求的优质品种培育成功。全基因组选择技术的应用也将加快畜禽遗传进展。然而现有育种技术主要使用DNA分子标记,RNA编辑或许可为畜禽育种开启新篇章。目前,在人的RNA编辑研究较为丰富并取得一定的成果,且针对中枢神经系统及癌症等疾病的RNA编辑研究表明,其在中枢神经系统发育及癌症发生发展中发挥重要作用,但在畜禽中关于RNA编辑的研究较为罕见,仅搜索到几篇基于高通量测序技术进行RNA编辑研究的报道[15, 90-91],尚未揭示RNA编辑对畜禽经济性状的影响。RNA现已成为研究热点,转录组测序相关报道呈现井喷之势,大量测序数据等待深入挖掘。同时,得益于生物信息学的快速发展,在全基因组范围内准确鉴定RNA编辑事件已成为现实。以期RNA编辑将作为新型标记用于疾病诊断及药物研发;对于与经济性状相关的RNA编辑位点,可以利用编辑技术在DNA和RNA水平上进行定点定向定时编辑;还可以联合SNP和RNA编辑开展全基因组选择,进一步提高选择准确性。由此可见,RNA编辑在畜禽育种方面具有广阔的应用前景,但同时也面临着诸多挑战,如研究不充分、检测费用高、已知功能的编辑位点数量少、具有时空特异性等。如何将RNA编辑信息更好地应用于畜禽育种还需进一步研究探讨。随着现代生物技术和研究方法的不断发展以及科学研究的持续深入,RNA编辑检测将会如SNP分析般常规,越来越多RNA编辑位点的功能也将得到揭示,以期鉴别更多与畜禽经济性状相关的基因位点,为畜禽育种提供更多的分子标记,促进相关产业的发展。



