李燕方,古 江,袁 媛,谢 跃,赖为民,杨光友,古小彬
(四川农业大学动物医学院,成都 611130)
中国是养兔大国,每年的兔肉、兔毛和兔皮产量均居世界前茅[1-2]。痒螨病是危害养兔业的一种常见外寄生虫病,我国家兔的感染率可达70%左右,该病是由绵羊痒螨(兔亚种)(Psoroptesovisvar.cuniculi)寄生于兔的外耳道所引起的一种以耳道内形成卷纸样结痂、瘙痒、病变部皮肤增厚为临床特征,如不及时治疗,甚至可引起死亡,每年给养兔业造成巨大的经济损失[3-5]。
微进化(又名内进化,microevolution)指种内或近源物种间的进化,是生物多样性源头和适应环境的基础。寄生虫的微进化可受其本身的生物学特性、宿主的种类和分布、所生活环境变化等因素的影响,且寄生虫的微进化对寄生虫种群的遗传多样性有重要意义[6-7]。通过研究遗传多样性可分析物种的进化潜力及应对复杂环境的能力及历史发展动态,推测未来发展趋势[8]。我国家兔饲养区域广泛,饲养家兔品种较多,且各饲养区处于不同的地理环境和温度带,这些因素对兔体表寄生的痒螨的影响尚不明确。因此,笔者采集我国5个区域的獭兔和肉兔源的痒螨虫株,选用线粒体12S基因为目的片段,分析不同区域来源和不同兔品种来源的痒螨线粒体12S基因的差异,从而探讨我国绵羊痒螨(兔亚种)的遗传多样性。
1 材料与方法
1.1 绵羊痒螨(兔亚种)样品的来源
1.1.1 样品采集地点 从我国华北地区(河北)、华东地区(山东)、华中地区(湖北、河南)、西北地区(陕西)和西南地区(四川、云南)自然感染绵羊痒螨病的獭兔和肉兔耳部采集获得89个痒螨虫株(表1)。
表1我国绵羊痒螨(兔亚种)不同种群样品采集情况表
Table1SamplingsitesofPsoroptesovisvar.cuniculipopulationsinChina
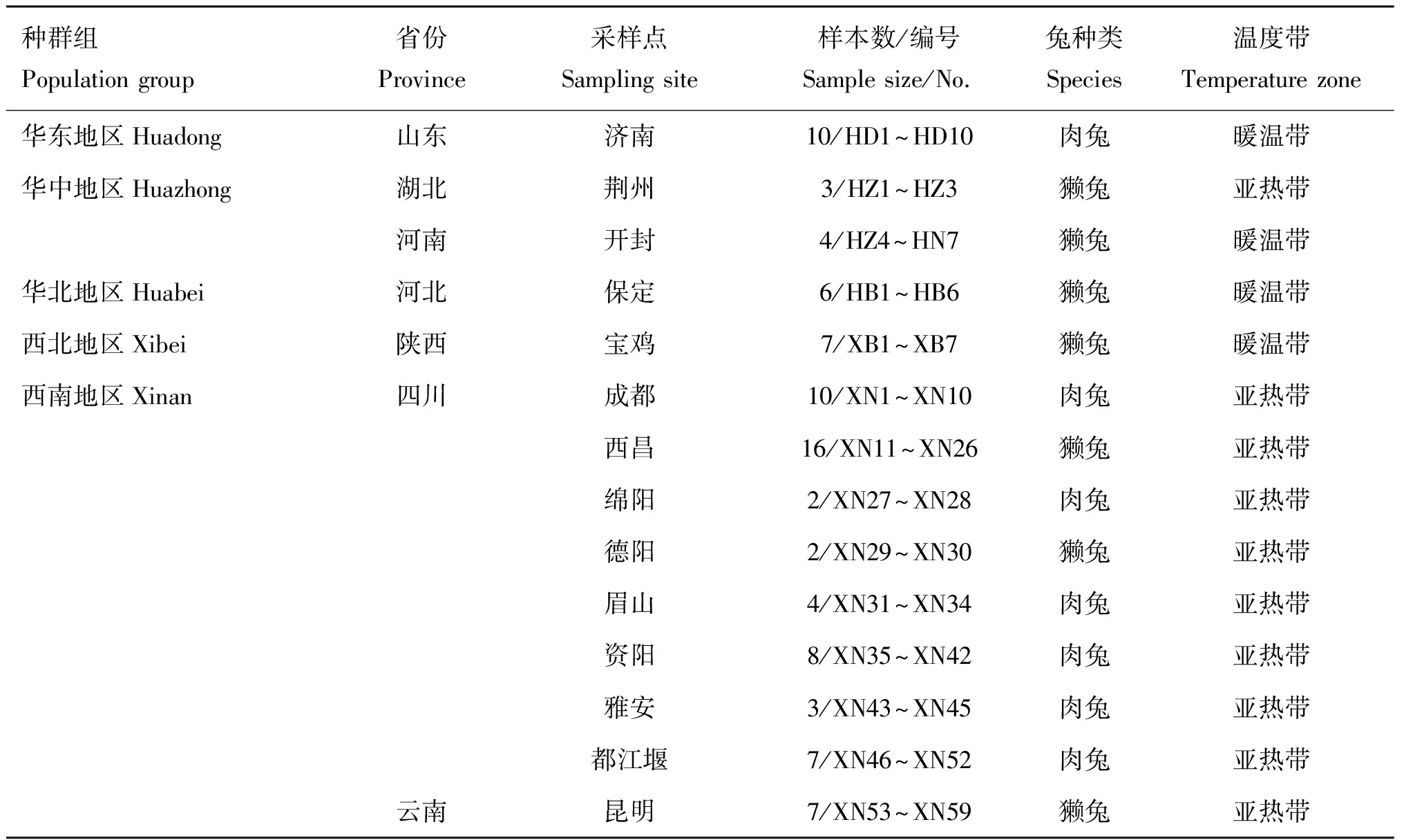
种群组Population group省份Province采样点Sampling site样本数/编号Sample size/No.兔种类Species温度带Temperature zone华东地区Huadong山东济南10/HD1~HD10肉兔暖温带华中地区Huazhong湖北荆州3/HZ1~HZ3獭兔亚热带河南开封4/HZ4~HN7獭兔暖温带华北地区Huabei河北保定6/HB1~HB6獭兔暖温带西北地区Xibei陕西宝鸡7/XB1~XB7獭兔暖温带西南地区Xinan四川成都10/XN1~XN10肉兔亚热带西昌16/XN11~XN26獭兔亚热带绵阳2/XN27~XN28肉兔亚热带德阳2/XN29~XN30獭兔亚热带眉山4/XN31~XN34肉兔亚热带资阳8/XN35~XN42肉兔亚热带雅安3/XN43~XN45肉兔亚热带都江堰7/XN46~XN52肉兔亚热带云南昆明7/XN53~XN59獭兔亚热带
1.1.2 痒螨的收集 将病兔耳部的痂皮用镊子轻轻取出,放入洁净的培养皿中,置于37 ℃培养箱中,30 min后除去皮屑,收集虫体,进行形态学鉴定[9]确认后-20 ℃保存。
1.2 引物的设计
根据GenBank绵羊痒螨(兔亚种)的线粒体基因组(GenBank No.:KJ957822)找到线粒体12S基因全序列,利用Primer Premier 5.0设计用于扩增12S全序列的特异性引物。引物序列如下,12S-F:5′-GCTTTGGGGGCTGTAGAATCAC-3′;12S-R:5′-CAAAAACCCTAAACAAGAGGCACC-3′。引物由上海生工生物科技公司合成。
1.3 单个虫体DNA的提取
将保存于-20 ℃的绵羊痒螨取出,每个样品挑出一只虫体放入一个EP管中,用尖头玻璃棒迅速研碎。然后采用OMEGA公司的Mollusc/Arthropod基因组DNA抽提试剂盒提取单个痒螨虫体的基因组DNA,保存于-20 ℃冰箱中待用。
1.4 PCR扩增及测序
25 μL的扩增体系包括PCR Mixture 12.5 μL,上下游引物各1.0 μL,模板DNA 4.0 μL,双蒸水6.5 μL。PCR反应条件:94 ℃预变性5 min,94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min 30 s,72 ℃ 延伸10 min。扩增结束将PCR产物于1.0%琼脂糖凝胶中电泳检查结果。将凝胶中条带大小与预期相符的胶块切割下来,移入1.5 mL灭菌离心管中,利用天根普通琼脂糖凝胶DNA回收试剂盒进行胶回收。将胶回收产物连接入pMD19-T载体,转化DH5α感受态细胞,蓝白斑筛选出阳性克隆,送上海生工生物科技公司进行正反向测序。
1.5 序列分析
1.5.1 遗传多样性分析 将测定的核酸序列运用DNAman 5.2对所测序列进行拼接、比对[10],用DNAStar 5.1对测序成功序列进行剪切修饰[11]。通过MEGA5.1软件计算平均核苷酸差异指数(average number of nucleotide difference,K)、核酸分歧度(nucleotide divergence,Dxy)、遗传距离[12]。运用Dna SP 5.1软件计算单倍型多样性(haplotypes diversity,Hd)、核苷酸多样性(nucleotide diversity,π)、单倍型数(number of haplotypes)[13]。采用Arlequin 3.1通过分子方差分析(AMOVA)计算各个种群间的遗传差异指数(Fst),基因流(Nm)通过Nm=1/4(1/Fst-1)计算获得[14]。
1.5.2 遗传结构分析 中性检验(neutrality tests)结果(Tajima’sDtest,Fu’sFstest)由Arlequin 3.1软件计算得到[14],结合Dna SP 5.1软件分析得到的错配分布曲线[13],以了解我国绵羊痒螨(兔亚种)的历史动态。采用MEGA 5.1软件构建各种群间的系统发育NJ树[12],并bootstraps 1 000次,检验聚类树的置信度;同时结合Network 5.0制作单倍型网络图[15]。
2 结 果
2.1 痒螨12S基因全序列的碱基组成及变异
成功获得了89个绵羊痒螨兔亚种样品的12S序列(登录号:MH361034~MH361122),89条12S基因全序列长度均为657 bp,其中A、T、C、G平均含量为31.34%、40.03%、10.20%、18.43%,平均A+T含量71.37%,平均C+G含量28.63%,出现明显的偏倚。89条12S基因序列共存在变异位点76个,其中多变异位点7个,单变异位点69个。
序列的突变方式大部分是碱基转换(转换/颠换= 6.9),其中包括90个转换(T↔C,A↔G),13个 颠换(G↔T,A↔T,A↔C,G→C),并且没有发现碱基的缺失。G→C颠换只存在于华中种群,华北种群和西北种群无颠换突变。所有种群中T↔C转换率比A↔G转换率高。
2.2 5个地理种群遗传多样性参数及中性检验值分析
用Dna SP 5.1软件分析发现获得的89条12S序列共检测出50个单倍基因型(H1~H50);其中,西南种群所含单倍型数最多(35个),有32个特有单倍型(H19~H50);其次为华东种群,所含单倍型数为8个,存在7个特有单倍型(H2~H8);华中和华北种群所含单倍型数相同,均为6个,分别存在特有单倍型1个(H12)和4个(H13~H16);以西北种群所含单倍型数最少(5个),存在2个特有单倍型(H17、H18)(表2)。50个单倍型中存在4个共享单倍型(H1、H3、H9和H11);其中H9和H11被西南与华中、华北、西北种群所共有,共享频率最高,分别为19.10%(17/89)和14.61%(13/89);H1被西南与华中、华东、西北种群共享,共享频率为12.36%(11/89);单倍型H3仅被华中和华东种群所共享,共享率2.25%(2/89)。
我国5大地区绵羊痒螨(兔亚种)种群的单倍型多样性(Hd)较高,范围为0.857 14(西北种群)~1.000 00 (华北种群),我国整体种群单倍型多样性(Hd)为0.931 05,表明我国种群单倍型多样性较为丰富。5个地区种群中,华北种群的核苷酸多样性(π)最低(0.003 55),华东种群的核苷酸多样性(π)最高(0.005 82),中国整体核苷酸多样性(π)为0.005 59 (表1),表明中国绵羊痒螨种群核苷酸多样性较高。我国5大地区种群的平均核苷酸差异指数(K)范围为2.333 33(华北种群)~3.822 22(华东种群)。
利用Tajima’sD和Fu’sFs检验得到的中性检测结果见表2。西南种群的Tajima’sD和Fu’sFs均呈负值,且在我国5个种群中绝对值最高(Tajima’sD=-2.406 94;Fu’sFs=-26.028 89)。华北种群与西南种群Fu’sFs为负值(-3.926 53和-26.028 89),且差异极显着,表明这两个种群近期可能经历过群体扩张;但我国绵羊痒螨整体种群的Tajima’sD(-0.928 31)和Fu’sFs(-7.066 71)检验结果均为负值且不显着,表明我国绵羊痒螨群体符合中性进化假说,虽然西南种群与华北种群近期有过群体扩张,但并未对我国整体绵羊痒螨种群的扩张有明显的影响,从而我国整体种群呈现较为稳定状态。
2.3 遗传距离、核酸分歧度及分子变异分析
从表3可见,我国绵羊痒螨的遗传距离范围值为0.003 97~0.006 54,其中华北地区与华东地区的遗传距离最大(0.006 54);华北地区与西北地区遗传距离最小(0.003 97)。我国绵羊痒螨的核酸分歧度(Dxy)范围为0.003 95~0.006 49,其中华北地区与华东地区、西北地区的核酸分歧度分别为0.006 49 和0.003 95。西南地区与其他4个地区相比,与西北地区之间的核酸分歧度(0.005 02)和遗传距离(0.005 06)均最小。
我国各种群间的基因流(Nm)为0.724 32至无限大(∞),遗传分化指数(Fst)为-0.094 25~0.256 59(表4),5个地区之间的基因交流情况有所不同。其中,西南与华中和西北种群之间的基因交流最为频繁,从而降低了西南与华中、西北种群间的核酸分歧度。我国整体绵羊痒螨种群的Nm和Fst分别为5.657 372和0.042 322, 表明我国绵羊痒螨整体基因流水平较高,遗传分化程度弱。分子方差分析结果发现,种群内和种群间的遗传变异方差比率分别为95.77%和4.23%,表明本研究中绵羊痒螨的遗传变异主要发生在种群内部。
表2绵羊痒螨(兔亚种)群体遗传多样性参数
Table2GeneticdiversityindexesofPsoroptesovisvar.cuniculi
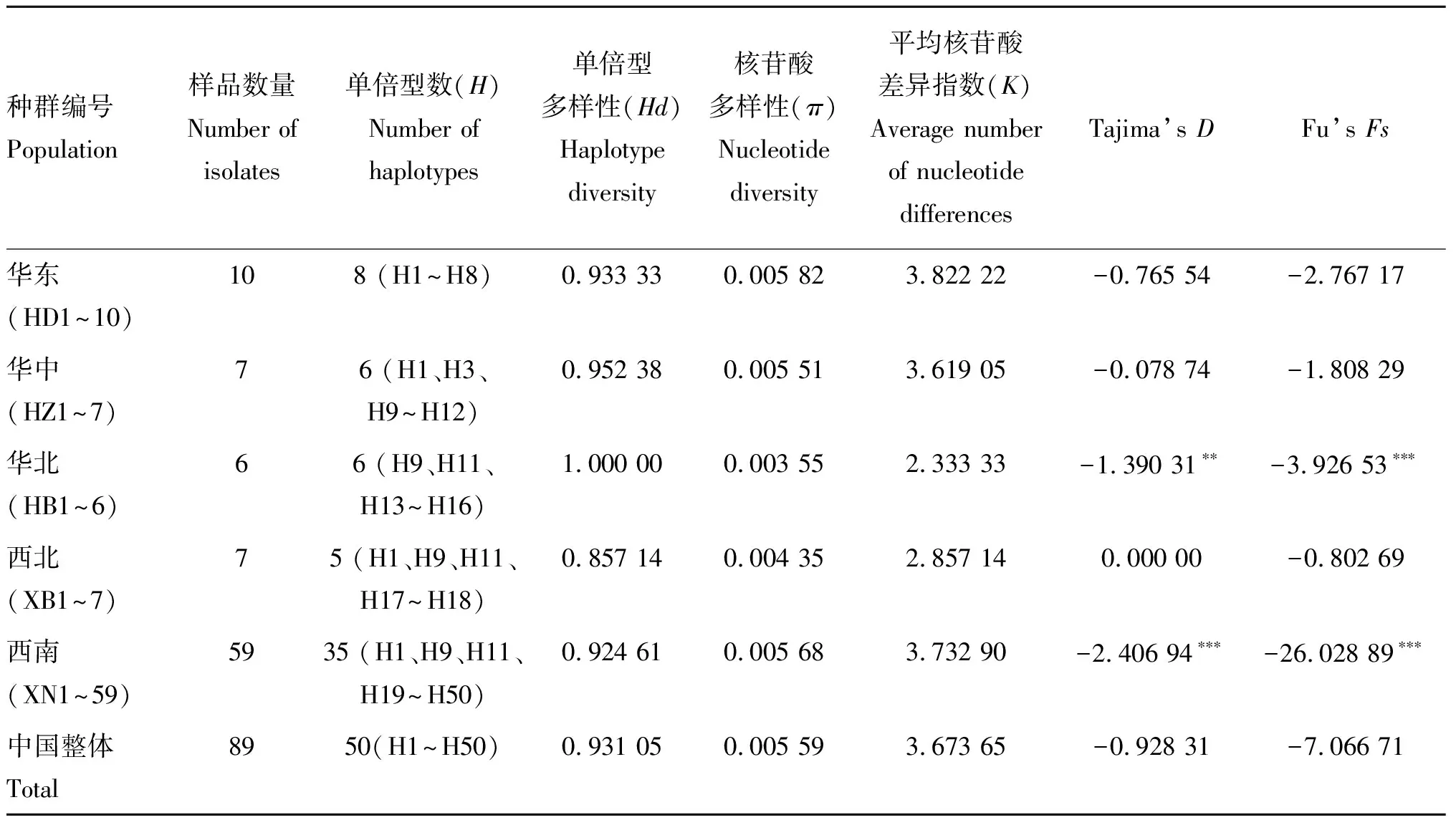
种群编号Population样品数量Number of isolates单倍型数(H)Number of haplotypes单倍型多样性(Hd)Haplotype diversity核苷酸多样性(π)Nucleotide diversity平均核苷酸差异指数(K)Average number of nucleotide differencesTajima’s DFu’s Fs华东(HD1~10)108 (H1~H8)0.933 330.005 823.822 22-0.765 54-2.767 17华中(HZ1~7)76 (H1、H3、H9~H12)0.952 380.005 513.619 05-0.078 74-1.808 29华北(HB1~6)66 (H9、H11、H13~H16)1.000 000.003 552.333 33-1.390 31∗∗-3.926 53∗∗∗西北(XB1~7)75 (H1、H9、H11、H17~H18)0.857 140.004 352.857 140.000 00-0.802 69西南(XN1~59)5935 (H1、H9、H11、H19~H50)0.924 610.005 683.732 90-2.406 94∗∗∗-26.028 89∗∗∗中国整体Total8950(H1~H50)0.931 050.005 593.673 65-0.928 31-7.066 71
*.P<0.05;**.P< 0.01;***.P<0.001
表3我国绵羊痒螨(兔亚种)各地理种群间的核酸分歧度(上三角)与遗传距离(下三角)
Table3Pairwisecomparisonsofthenucleotidedivergence(abovediagonal)andgeneticdistance(belowdiagonal)betweengeographicalpopulationsofPsoroptesovisvar.cuniculi
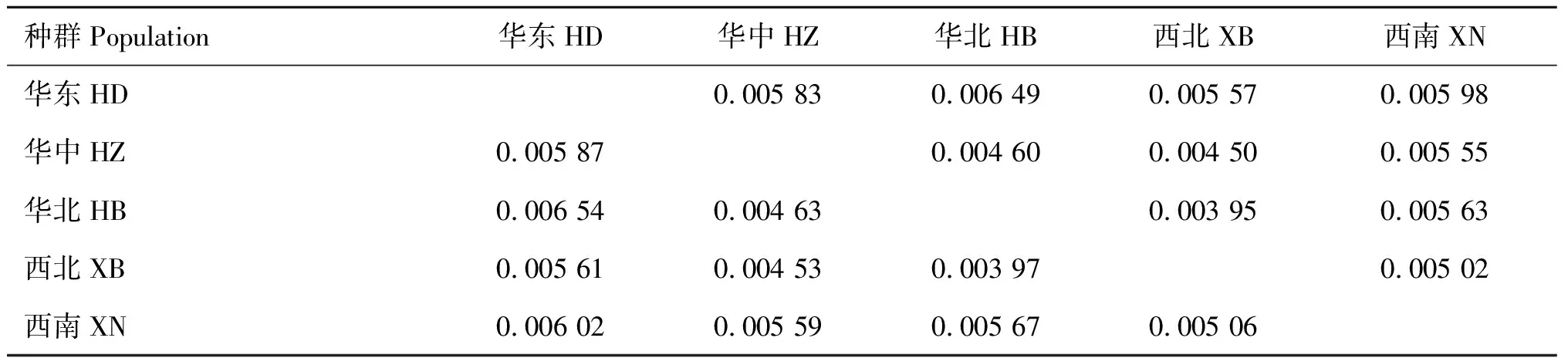
种群Population华东HD华中HZ华北HB西北XB西南XN华东HD0.005 830.006 490.005 570.005 98华中HZ0.005 870.004 600.004 500.005 55华北HB0.006 540.004 630.003 950.005 63西北XB0.005 610.004 530.003 970.005 02西南XN0.006 020.005 590.005 670.005 06
表4不同绵羊痒螨(兔亚种)种群间的基因流(Nm)(上三角)和遗传分化度(Fst)(下三角)
Table4Geneflow(Nm) (abovediagonal)andpopulationpairwisefixationindex(Fst)values(belowdiagonal)amongdifferentPsoroptesovisvar.cuniculipopulations
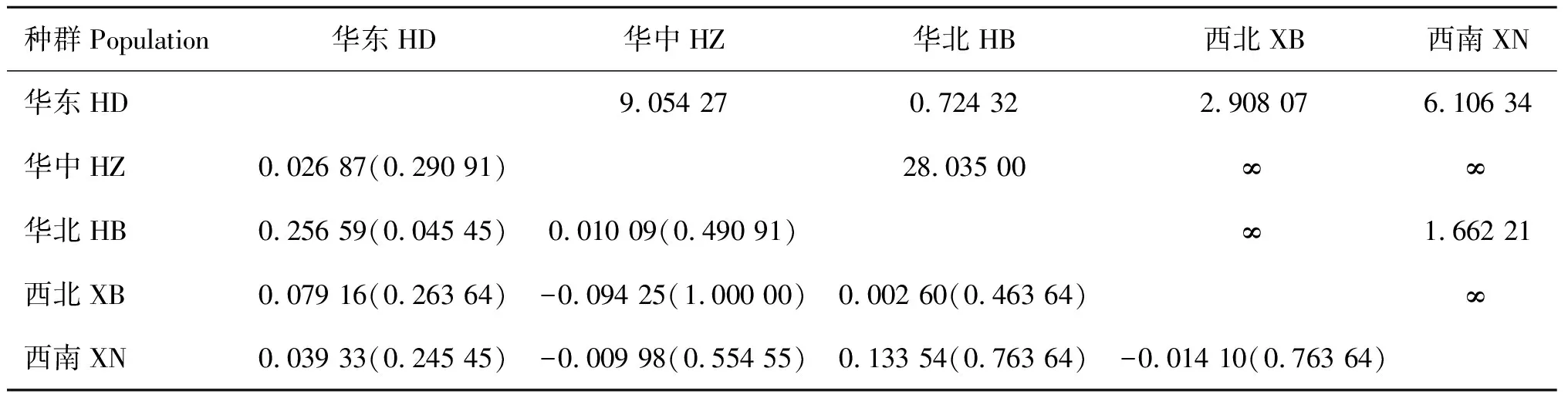
种群Population华东HD华中HZ华北HB西北XB西南XN华东HD9.054 270.724 322.908 076.106 34华中HZ0.026 87(0.290 91)28.035 00∞∞华北HB0.256 59(0.045 45)0.010 09(0.490 91)∞1.662 21西北XB0.079 16(0.263 64)-0.094 25(1.000 00)0.002 60(0.463 64)∞西南XN0.039 33(0.245 45)-0.009 98(0.554 55)0.133 54(0.763 64)-0.014 10(0.763 64)
2.4 系统发育树的构建及种群整体遗传结构分析
以疥螨(Sarcoptesscabiei)的12S基因(GenBank No.: NC_031334)为外群,利用MEGA 5.1软件采用NJ法构建分子系统树,结果显示绵羊痒螨5个地理种群的50个单倍型散在分布于不同的支系内,如西南种群的35个单倍型散在于不同的分支(图1),未按兔品种、温度带和地域来源的不同而聚类,而呈现为混杂的分布格局(图1)。
为了确定单倍型的谱系关系,采用Network 5.0软件以MJ(Median-Joining network)法构建5个不同地理种的50个单倍型(包括共享单倍型和特有单倍型)的网络图(图2),单倍型网络图具有丰富的分支和节点,以优势单倍型(H1、H9、H11)为中心呈辐射状分布,缺失3个单倍型(图2中红色圆圈,分别用mv1~mv3表示),且各单倍型的聚类情况与NJ树类似,并未按兔品种、温度带和地域来源进行聚类。对我国绵羊痒螨世系进行群体历史动态分析,核苷酸不配对分布呈双峰型(图3),表明早前未曾发生过群体扩张,长期处于一个较为稳定的状态。
3 讨 论
寄生虫种群遗传多样性会受地理环境[16]、寄生虫生物学特性、与宿主间的互相适应等多种因素影响[17]。我国兔业养殖历史悠久,家兔养殖分布于除西藏外的各省、市、自治区[1-2],这些不同地理区域包含了山地、高原、丘陵、盆地、平原等形态,地势地貌差异较大。这些差异较大地势地貌对绵羊痒螨兔亚种种群是否存在较大影响尚不明确。目前,绵羊痒螨种群的遗传多样性研究较为缺乏,仅笔者课题组的赵习彬等[18]利用线粒体细胞色素氧化酶b(Cytb)基因全序列进行了相关研究,12S基因作为一种常见的分子标记,进化速度与mtDNA基因组的平均进化水平基本相同[19],已广泛用于研究动物寄生性蜱螨种群遗传学[20-21]。目前,GenBank中除笔者课题组登陆的痒螨线粒体基因全序列中包含有12S序列信息外[22],尚未见痒螨的其他12S序列,因此本研究基于我们测序得到的89条绵羊痒螨12S基因全序列探讨我国绵羊痒螨兔亚种种群的遗传变异,以期为进一步明确我国绵羊痒螨兔亚种的遗传多样性提供基础。
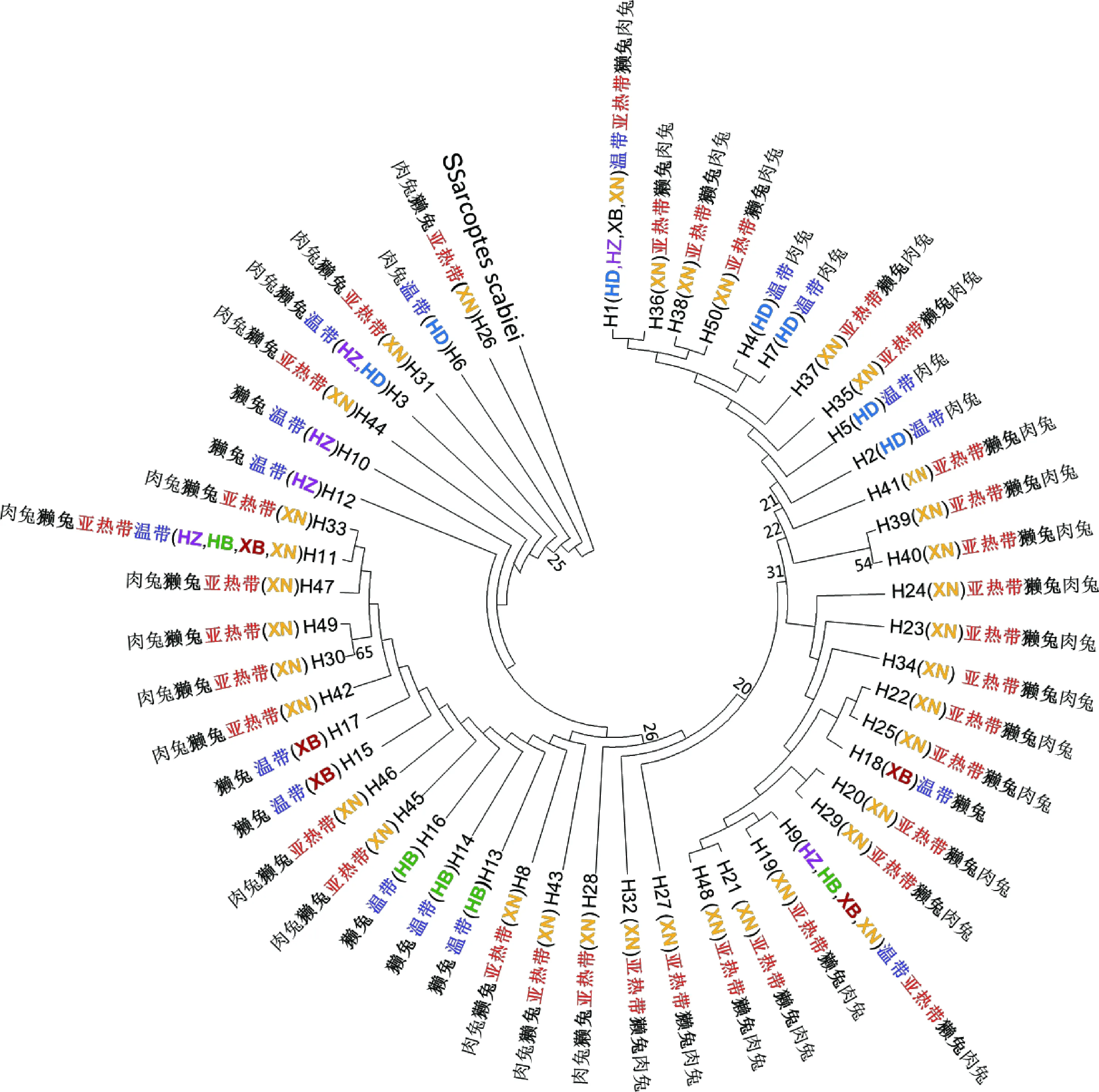
H1~H50表示50个单倍型;XN、HD、HZ、HB、XB分别表示西南、华东、华中、华北、西北
H1-H50 represent 50 haplotypes; XN, HD, HZ, HB, XB, represent Xinan, Huadong, Huazhong, Huabei, Xibei
图1 基于50个绵羊痒螨12S基因单倍型构建的NJ树
Fig.1 Neighbour-joining tree constructed from the 50 haplotypes of mtDNA 12S gene in Psoroptes ovis var. cuniculi based on Kimura-2-parameter distances
赵习彬等[18]基于Cytb全基因发现我国绵羊痒螨(兔亚种)种群中存在着丰富的核苷酸多样性(0.012 0)和单倍型多样性(0.959 2),比笔者基于线粒体12S基因全序列得到的核苷酸多样性(0.005 59)和单倍型多样性(0.931 05)高,可能与Cytb基因(1 100 bp)全序列的长度大于12S基因全序列(657 bp),其所含有的信息位点更多有一定的关系;综合Cytb和12S基因的核苷酸多样性和单倍型多样性来看,两者值均较高,表明我国绵羊痒螨(兔亚种)种群有较高的遗传多样性水平。我国痒螨种群呈现的这种较高水平的核苷酸多样性和单倍型多样性,将更利于痒螨在进化过程中通过产生多种变异来应对不良环境因素[23],有史可寻中国地区发生过一些如旱灾、雪灾等灾害,而这种遗传变异可能在痒螨抵御这些不利环境因素中起到重要作用[24]。赵习彬等[18]和笔者本次研究的群体历史动态分析结果均呈现我国绵羊痒螨种群整体的中性检测Tajima’sD和Fu’sFs检验结果均为负值,且差异均不显着,说明种群在历史演变中积累较多基因变异[25]。结合错配分布曲线(图3)中出现的两个峰值,表明绵羊痒螨种群在长期的历史进化过程中一直处于一个较为稳定的状态,种群早期可能并未经历过较大的群体扩张[26],由此可见,即使个别种群(华北和西南)发生过群体扩张过程,使整体种群表现出较弱的突然增长,但并未影响到种群的整体稳定,这与赵习彬等[18]的研究结果类似。
地理距离是影响种群间基因交流的重要因素之一[27],但赵习彬等[18]和笔者的研究发现绵羊痒螨(兔亚种)各地理种群间的基因交流与地理距离的远近并未全部表现出正相关的联系,基于绵羊痒螨Cytb基因发现,山东和河北的地理距离最近(340 km),但山东和河北两地的绵羊痒螨种群间的遗传距离不是最小的(0.001);本次研究基于12S基因发现保定(华北种群代表)与宝鸡(西北种群代表)间的地理距离比保定与济南(华东种群)的距离远,但华北、西北种群内的基因交流值(趋于无穷大)却大于华北、华东种群(0.724 32)。
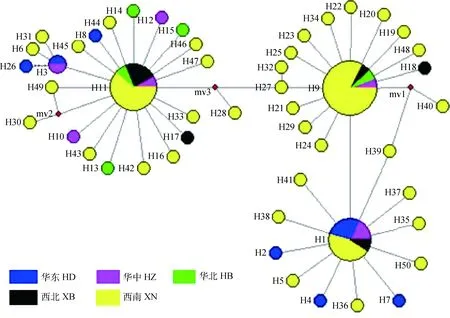
圆圈的大小与单倍型的分布频率成正比;不同颜色代表不同样品来源
The circle sizes are proportional to the number ofisolates;Different colors represent different sample sources
图2 基于50个绵羊痒螨12S基因单倍型单倍型构建的单倍型网络图
Fig.2 Network of mtDNA 50 12S gene haplotypes in Psoroptes ovis var. cuniculi

图3 基于12S gene序列分析我国89株绵羊痒螨(兔亚种)虫株的错配分布曲线
Fig.3 The mismatch-distribution of 89 Psoroptes ovis var. cuniculi isolates based on 12S gene sequence
由此可见,较远的地理隔离不能阻碍绵羊痒螨种群间频繁的基因交流,而这种基因交流单靠痒螨自身是不可能完成的,因为痒螨虫体较小,大小约(0.3~0.9) mm ×(0.2~0.52) mm,其爬行能力较弱[9],而不同地区间的兔贸易为各地痒螨种群间的基因交流提供了机会。然而,早期我国交通不便,较远地区间的兔贸易较少,导致各地痒螨种群间的基因交流较少;自20世纪50年代以来,中国家兔饲养量急剧增加[1],交通便捷,各地间的兔贸易日益频繁,在一定程度上增加了各地的痒螨种群之间的基因交流。
Fst是衡量种群遗传分化的指标,当0.00≤Fst< 0.05时,遗传分化程度很弱;当0.05≤Fst<0.15 时遗传分化程度中等; 0.15≤Fst≤0.25时遗传分化程度较大[28]。基于12S基因分析得到的Fst值(Fst=0.042 322)发现我国的绵羊痒螨种群存在一定的遗传分化,从而导致各地理种群间存在多个特有单倍型(46/50);但绵羊痒螨种群分化程度较弱(Fst<0.05),这与种群间频繁的基因交流相关(Nm=5.657 372);绵羊痒螨种群的这种遗传分化可能是由地理环境差异、历史自然灾害、兔的品种等因素所造成的,但这种遗传分化可能由于时间不够尚未累积形成新的种群遗传结构,这点在NJ树和单倍型网络图分析中得到证实(图1、图2)。
单倍型网络图显示出优势单倍型有3个(H9、H11、H1)(图2),有研究说明优势单倍型往往具有更加广泛的地理分布[29-30],笔者的研究亦证实了这点,其中优势单倍型H9、H11、H1广泛分布在华东、华中、华北、西北、西南5个地区。另外单倍型网络图(图2)中不能直接看出单倍型之间的进化关系,但出现历史单倍型的缺失(图2中红色圆圈,分别用mv1~mv3表示),这可能是因为某些地区样本数量太少,并且各单倍型的聚类情况与NJ树类似(图1),总体上看,没有明显的地域性差别,也并未按兔品种、温度带进行划分,这点聚类特点在赵习彬等[18]基于线粒体Cytb基因的研究中亦得到证实。
4 结 论
经对华东、华中、华北、西北、西南5个地区绵羊痒螨线粒体12S基因进行分析,发现我国绵羊痒螨(兔亚种)单倍型多样性高,核苷酸多样性低,基因交流频繁,未形成以兔品种、温度带和地域来源划分的种群遗传结构。



