米热姑丽·麦麦提,周世玉,刘 鹏,孟 阳,聂文悦,滕培晨,单文娟
(新疆大学生命科学与技术学院, 新疆生物资源基因工程重点实验室,乌鲁木齐 830046)
托氏兔(LepustolaiPallas,1778)又称蒙古兔,隶属于兔形目(Lagomorpha)兔科(Leporidae)兔属(LepusLinnaeus, 1758),体型较大,迁移能力强,分布范围广,可生活在多种环境中,在我国东北部、中部及西北部地区均有分布[1]。由于野兔种间形态、外形、头骨和牙齿等方面差异较小,而且野兔间存在杂交现象,给分类工作带来了很多困难,托氏兔曾被包括在草兔(L.capensis)或欧兔(L.europaeus)中[2]。罗泽珣[3]对比了我国“草兔”和欧兔的外形和头骨等特征,发现有明显差别,否定了我国“草兔”归属于欧兔的观点。基于分子生物学[4-5]和头骨测量数据[6]的研究不支持将我国“草兔”归为L.capensis, 因此将新疆原“草兔”种群划分为托氏兔和藏兔(L.tibetanus),其中天山以北的种群为托氏兔[7]。近年来的研究显示,托氏兔在新疆分布有两个亚种,其中分布在新疆北部和西北部地区的野兔为西域亚种(L.tolailehmanni),分布在新疆中部和东部地区的野兔为中亚亚种(L.tolaicentrasiaticus)[7-8]。
早期关于托氏兔分子生物学方面的报道将托氏兔误分成草兔亚种进行研究,如Wu等[4]与Wang和Yang[5]研究了不同野兔类群间的系统发育关系,其中包括新疆北部及中部的野兔;Liu等[9]基于4个线粒体DNA(mitochondrial DNA,mtDNA)片段和核基因,首次证明了6个中国兔属物种之间通过历史和近期的种间杂交发生了频繁的基因渐渗事件,其中包括分布在新疆北部的野兔;Wu等[10]发现,塔里木兔(L.yarkandensis)和新疆“草兔”的杂交个体中存在广泛的双向线粒体DNA和SRY基因渐渗;单文娟等[11]基于D-LOOP探究新疆“草兔”的种群遗传结构和亚种分化,结果显示,新疆“草兔”具有较高的遗传多样性和明显的系统地理结构。近年来,关于新疆托氏兔的研究集中在测定线粒体基因组全长[12]、筛选DNA条形码[13]、鉴定野兔性别[14]及年龄[15]等方面。除此之外,还有基于线粒体基因评价新疆3种野兔遗传多样性方面的报道[8],该研究中包含新疆北部阿勒泰与中部达坂城和托克逊的少量托氏兔样本,缺乏广泛的取样。
野兔由于繁殖能力和适应能力强,除个别种类外,多数种类的种群数量相对较多,相关研究较少。目前,对新疆托氏兔的研究主要集中在分类现状与分布,而关于分子生物学方面的研究大多是在托氏兔分类有误时进行的,还未见较全面地评价新疆托氏兔种内遗传多样性水平,探究亚种间遗传结构等方面的报道。遗传多样性及种群遗传结构研究是种质资源开发与利用的一项重要内容。野兔主要以草本植物和农作物为食,对农林牧业有一定的伤害,而且兔肉和皮毛具有一定的经济价值[16]。2009年,常卫利和兰文旭[17]报道,新疆阿勒泰地区托氏兔数量激增,威胁到当地农业生产工作,因此当地实行了限量捕猎利用托氏兔。目前,新疆托氏兔的遗传多样性现状是未知的,因此本研究采用CO1和D-LOOP基因,结合系统发育和群体遗传学的分析方法,评价新疆托氏兔的遗传多样性水平,并探究亚种间的遗传结构,旨在揭示新疆托氏兔的遗传潜力和环境适应能力,为托氏兔的保护和利用提供分子遗传学方面的依据。
1 材料与方法
1.1 样本收集及采样点区域划分
本研究共收集了78个新疆托氏兔的样本,分别采自新疆12个地区(表1)。为方便分析,根据样本地理分布将本研究中的托氏兔样本分为4个地理组群,分别为北部、西北部、中部以及东部组群,见表1。
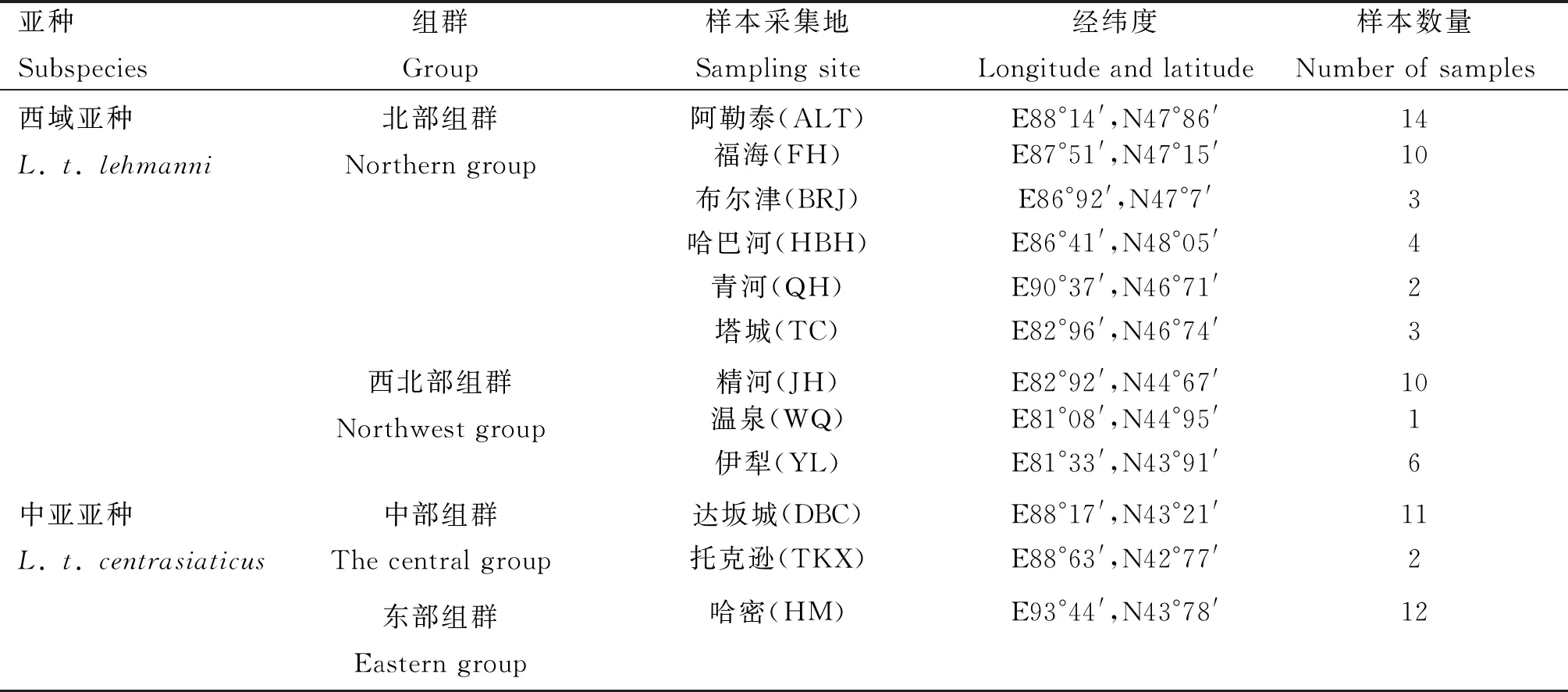
表1 新疆托氏兔样本采集信息
1.2 基因组DNA提取、PCR扩增
采用DNA组织提取试剂盒提取样本总DNA,对提取质量较好的DNA产物进行PCR扩增。本研究采用mtDNA的CO1和D-LOOP基因,CO1的PCR引物序列[13]为F:5′-AGGAACAGCCCTYAGTCT-3′,R:5′-GGTGGGCTCAAACAATAA-3′;D-LOOP区的PCR引物序列[18]为F:5′-CAGAGATGGAGATYAACTC-3′,R:5′-GCATGG-GCTGATTAGTCAT-3′, PCR引物送至生工生物工程(上海)股份有限公司合成。CO1和D-LOOP基因的PCR扩增体系均为25 μL:Premix Taq(1.25 U·25 μL-1)13 μL,上、下游引物(10 μmol·L-1)各1 μL, DNA模板1 μL,无菌去离子水9 μL。扩增条件为:95 ℃变性3~5 min;94 ℃ 变性30 s,51 ℃退火30 s,72 ℃延伸60 s,25~35次循环;72 ℃延伸10 min。 PCR产物进行1.5%的琼脂糖凝胶电泳。电泳结束后通过凝胶成像系统成像,将扩增结果良好的PCR产物进行测序。
1.3 数据分析
用DNAMAN v6.0.40和Clustalx 1.83[19]处理序列格式;将2个基因的序列用MegaX[20]分别进行多重序列比对,比对完成后将序列减齐;用Phylo-Suite 1.2.2[21]将两个基因按照线粒体DNA上的顺序拼接起来。用Mega X和DnaSP 5[22]进行序列特征分析。用Arlequin v3.5[23]计算遗传多样性指数(genetic diversity index)和中性检验参数(neutrality tests),进行分子变异分析(analysis of molecular variance,AMOVA),计算各种群间的遗传分化指数(F-statistics,Fst),并进行错配分布分析(Mismatch distribution)。用MegaX构建ML树,用PhyloSuite v1.2.2构建贝叶斯树。用PopART[24]软件绘制单倍型中介网络图(median-joining network)。根据公式Nm=(1-Fst)/4Fst计算基因流大小[25]。用Beast v2.3.0[26]进行扩展贝叶斯天际线分析(Extended Bayesian Skyline Plots,EBSPs),Prior设置为Coalescent Extended Bayesian Skyline Plot。
2 结 果
2.1 基因组DNA提取与PCR扩增
从托氏兔肌肉组织中提取基因组DNA,以此为模板扩增mtDNA的CO1和D-LOOP片段。电泳检测得到与预期目标片段大小一致的清晰条带(图1), 可用于下一步的测序。
M. DNA相对分子质量标准;1~5. CO1和D-LOOP片段的PCR扩增产物M.DL1500 marker; 1-5. PCR products of CO1 and D-LOOP图1 2个mtDNA片段的部分PCR扩增结果电泳图Fig.1 The partial electrophoresis results of PCR amplification for 2 mtDNA fragments
2.2 新疆托氏兔mtDNA片段的序列特征
将mtDNA的CO1和D-LOOP基因序列拼接,得到总长为1 170 bp的合并序列。经分析其中T、C、A、G碱基平均含量分别为31.6%、28.2%、26.6%、13.6%。T+A含量为58.2%、C+G含量为41.8%,T+A含量明显大于C+G含量,存在A/T偏倚性。共计发现多态性位点(variable sites)192个,突变位点(mutations sites)217个,简约信息位点(parsimony informative sites)161个,其中转换(transition)43个、颠换(transversion)13个,插入/缺失(insertion/deletion)35个。
2.3 新疆托氏兔的遗传多样性
78个新疆托氏兔样本的合并序列共定义了68种 单倍型(表2)。托氏兔4个地理组群的单倍型多样性(haplotype diversity,h)为0.985~1.000;托氏兔4个组群中西北部组群的核苷酸多样性(nucleotide diversity,π)最高,为0.045±0.023,中部组群的核苷酸多样性最低,为0.029±0.015,托氏兔总的核苷酸多样性为0.049±0.023。
2.4 新疆托氏兔的系统发育关系
为探讨新疆托氏兔的系统发育关系,本研究以家兔(Oryctolaguscuniculus)为外群,基于CO1和D-LOOP基因合并的单倍型序列采用贝叶斯法和最大似然法构建系统发育树,得到两种树的拓扑结构基本一致。系统发育树结果显示(图2),本研究所用的托氏兔样本被分成置信度较高的3个进化枝,其中Clade A包含多数北部和少数西北部组群的样本;Clade B中绝大多数为中部和东部组群的样本,来自同一个组群的样本聚在一起;Clade C包含北部和西北部以及少数中部组群的样本。由此可见,虽然有部分样本混杂,但每个进化枝均包含了大多数来自相同或相邻区域的样本,样本的聚类情况与分布范围和地理距离相吻合,说明新疆托氏兔的分布具有比较明显的系统地理分布格局。
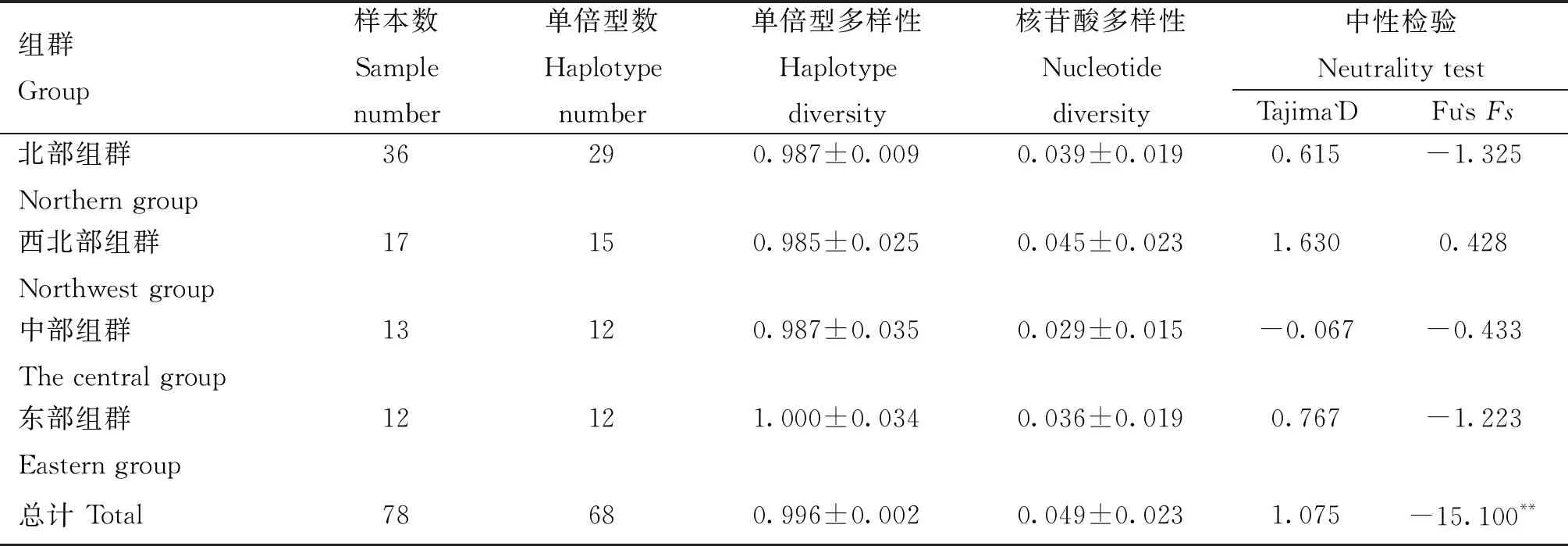
表2 基于mtDNA的CO1和D-LOOP基因计算的新疆托氏兔的遗传多样性和中性检验参数
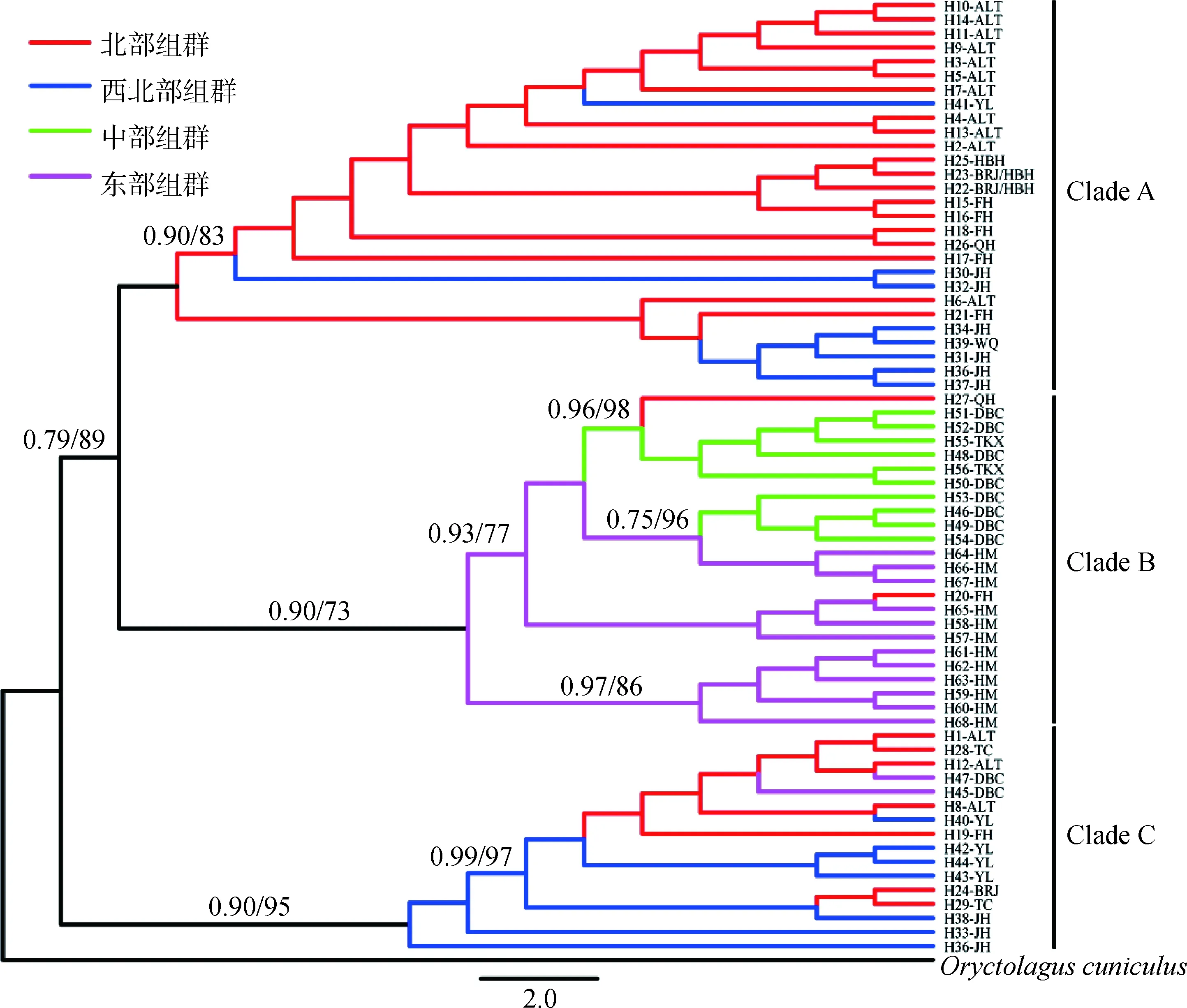
进化枝上的数字分别代表贝叶斯树置信度和ML树置信度Numbers on the branches represent Bayesian posterior probabilities and ML tree posterior probabilities图2 基于mtDNA的CO1和D-LOOP基因的单倍型序列构建的系统发育树Fig.2 Phylogenetic tree based on the haplotype sequence of CO1 and D-LOOP genes of mtDNA
2.5 新疆托氏兔的遗传结构
进一步以68个单倍型构建了中介网络图(图3),其结构与系统发育树一致。中介网络图也明显分为3个支系,其颜色标注与系统发育树一致。中介网络图中,左下方的一大枝包含多数北部和部分西北部组群的样本;右边的大枝分为两个小枝,分别为中部和东部组群的样本;左上枝包含北部、西北部以及少数中部组群的样本。中介网络图的结果也支持新疆托氏兔具有较明显的系统地理分布格局,除少量样本比较混杂,来自同域或邻域的样本聚在一起。同时,从中介网络图中可以看出,新疆托氏兔所有单倍型为各地理组群的特有单倍型,组群间没有共享单倍型,地理组群内也仅有2个相邻的地方共享的单倍型(H22和H23,图3未显示,可看图2),说明新疆托氏兔具有很高的单倍型多样性。
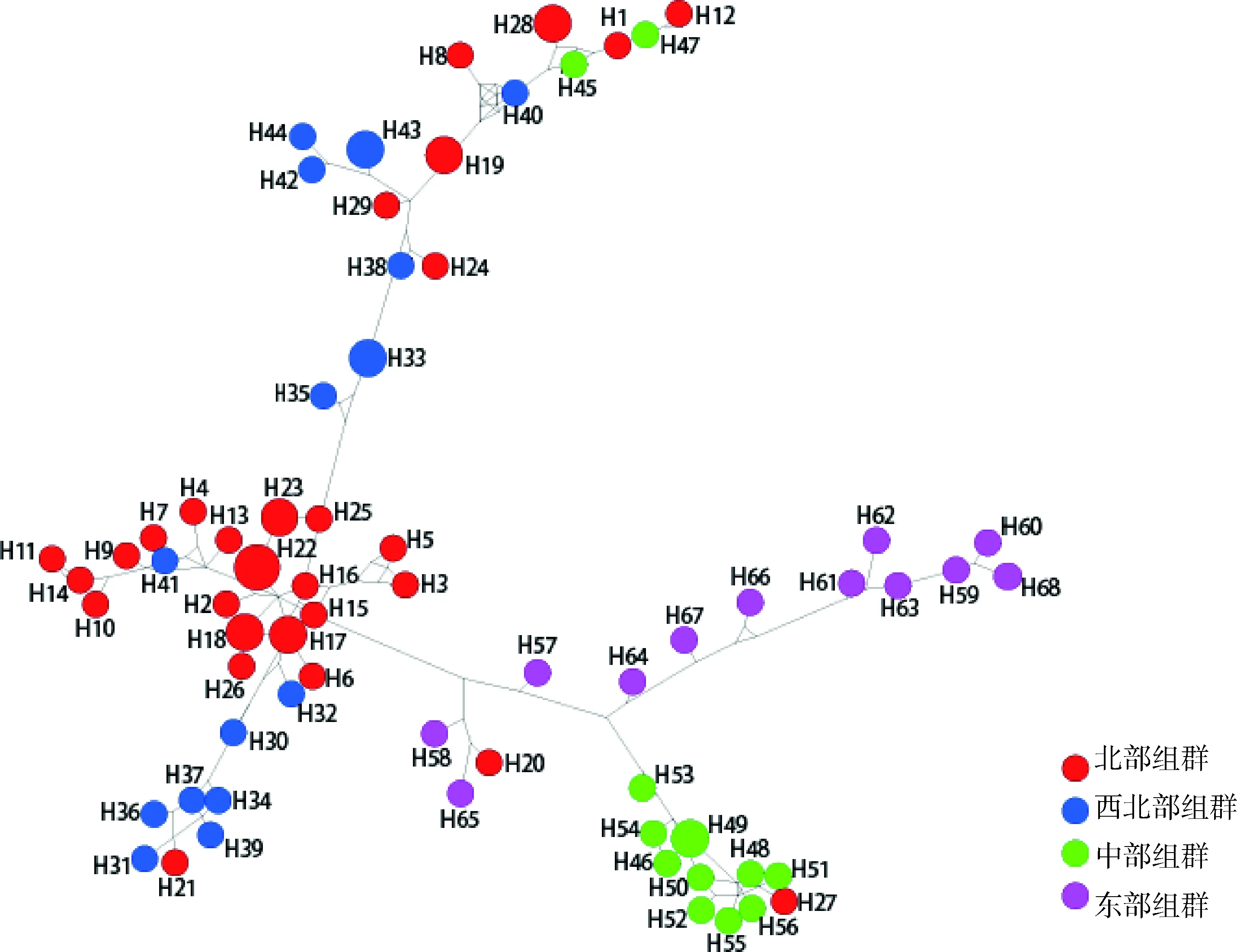
圆圈代表每个单倍型;圆的面积与单倍型频率成正比;直线距离代表突变步数;不同颜色表示不同的地理组群The circle represents each haplotype; The area of circle is proportional to the haplotype frequency; The linear distance represents the number of mutation steps; Different colors represent different geographic groups图3 新疆托氏兔68个单倍型的中介网络图Fig.3 The median-joining network of 68 haplotypes of L. tolai in Xinjiang
AMOVA分析结果显示(表3),将新疆托氏兔划分为4个不同地理组群时,组群内各种群间的变异占27.300%,且P值极显着(P=0.000<0.01),变异贡献率较小;将托氏兔按照西域亚种和中亚亚种分组时,种群内的变异占67.530%(P=0.000<0.01),组群间的变异大于组群内各种群间的变异,分别为19.400%、13.070%,且P值均极显着;将托氏兔按照西域亚种、中部和东部组群分组时,种群内的变异占67.560%,组群内各种群间的变异占8.260%,且P值均极显着。
种群间的遗传分化指数Fst值显示(表4),北部组群与西北部组群间的Fst值(为0.101,P<0.05)最小,中部组群与东部组群间的Fst值(为0.279,P<0.01)次之;托氏兔西域亚种与中亚亚种间的Fst值均较大(分别为0.331、0.306、0.376、0.340),且P值均极显着。基因流结果显示(表4),除了北部与西北部组群间的基因流较大、为2.225,其余组群间的基因流均较小。
2.6 新疆托氏兔的种群历史动态
本研究采用中性检验Tajima`D、Fu`sFs、核苷酸错配分布分析和EBSPs等方法分析新疆托氏兔的种群历史。中性检验结果显示(表2),4个地理组群的Tajima`D和Fu`sFs的绝对值均较小(0.067~1.630),P值均不显着(P>0.05),托氏兔总的Fu`sFs为-15.100(P=0.007)。核苷酸错配分布图呈现多峰(图4A)。扩展贝叶斯天际线(EBSPs)结果显示,在距今大约0.01 Ma(百万年)时,新疆托氏兔群体发生了扩张(图4B)。
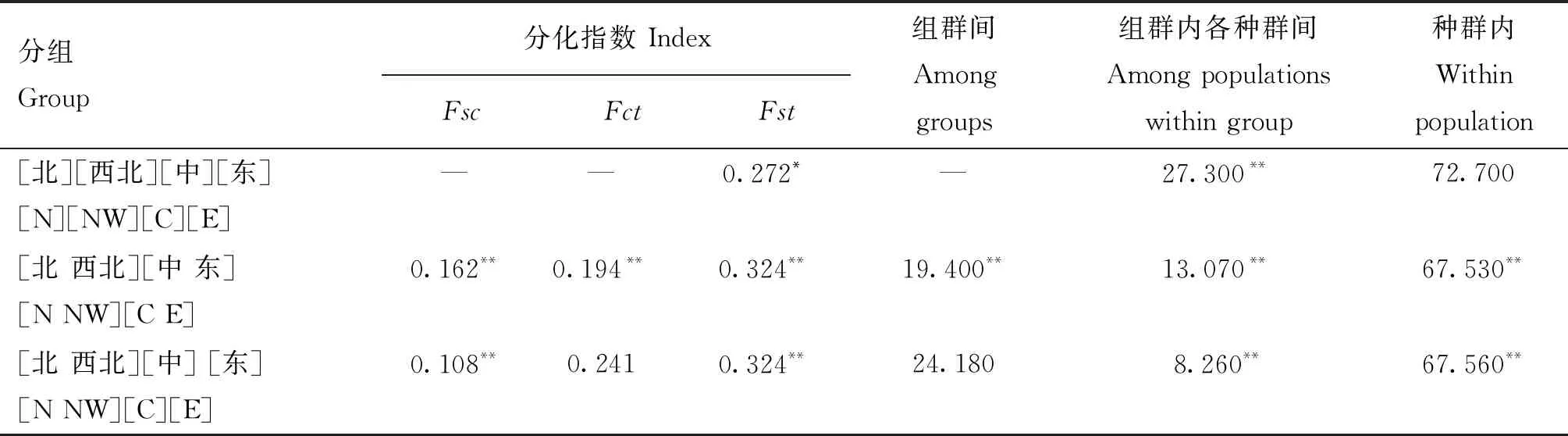
表3 新疆托氏兔mtDNA单倍型分子变异等级分析(AMOVA)
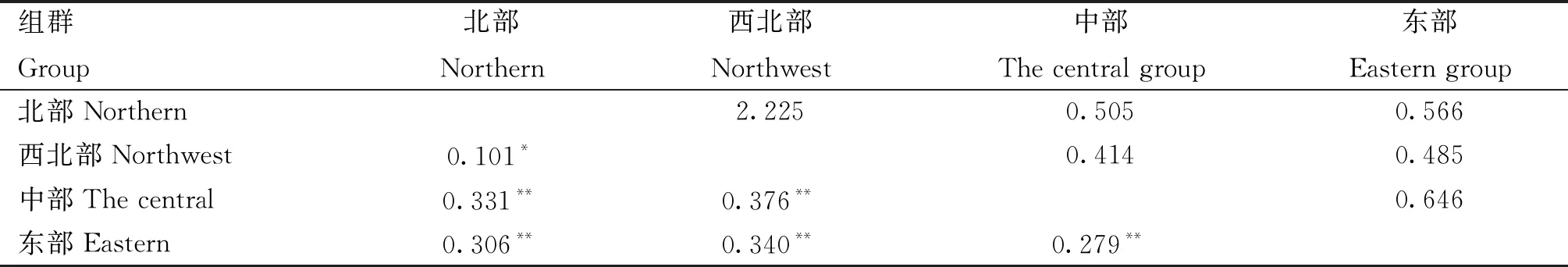
表4 新疆托氏兔4个地理组群间的遗传分化指数(对角线以下)及基因流(对角线以上)
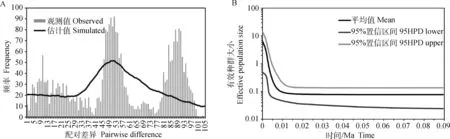
A. 核苷酸错配分布图;B. 扩展贝叶斯天际线图A. Mismatch distribution analysis of nucleotides; B. Extended Bayesian skyline plots analysis图4 新疆托氏兔的种群历史分析Fig.4 Population history analysis of L. tolai in Xinjiang
3 讨 论
3.1 新疆托氏兔的遗传多样性
遗传多样性的高低反映了物种的进化潜力和应对环境变化的能力,一个物种具有越高的遗传多样性,其应对环境变化的能力就越强。相比于其他的野兔,如欧兔(L.europaeus)h=(0.965±0.006)、π= (0.025±0.002)[27],藏兔(L.tibetanus)h=(0.972土0.064)、π=(0.002±0.001)[8],塔里木兔(L.yarkandensis)h=0.987[28],本研究检测到的新疆托氏兔遗传多样性水平较高(h=(0.996±0.002)、π=(0.049±0.023))。代慧英和单文娟[15]通过头骨形态指标鉴定新疆野兔年龄,其中包括新疆北部和中部的托氏兔样本,结果表明新疆野兔年龄结构稳定,种群繁殖能力较强,种群数量呈稳定增长模式;伊拉木江·托合塔洪等[29]对新疆野兔的生境分布研究显示,对于新疆托氏兔,当前气候条件下适宜生境分布在阿勒泰、塔城、伊宁以及哈密北部等地区,这些地区与目前新疆托氏兔的分布区域基本重合。综上,新疆托氏兔较高的遗传多样性可能是由于兔属物种长远的历史进化以及较大的种群数量导致了群体内遗传变异有效的保存和积累,同时较适宜的环境条件对此也做出了一定的贡献。
3.2 新疆托氏兔的遗传结构
本研究基于mtDNA的CO1和D-LOOP基因构建的系统发育树(图2)将新疆托氏兔群体分为置信度较高的3个支系,除个别样本聚类情况较为混杂,同域或邻近分布的野兔聚在一起,中介网络图的结果(图3)也与此相符,说明新疆托氏兔具有较明显的系统地理结构。本研究中,Clade A和C中既包含北部组群的样本又包含西北部组群的样本,同时这两个组群间的遗传分化相对较小(Fst为0.101),而且存在较强的基因流(2.225)。AMOVA分析也显示(表3),将北部组群和西北部组群分为一组时,两个组群间的变异贡献率很小(两种分析方法分别为13.070%和8.260%,P值均极显着),说明北部组群和西北部组群间分化很小。在形态分类学中,北部和西北部组群的野兔属于托氏兔西域亚种。新疆北部的阿勒泰地区和塔城地区与西北部的精河、温泉及伊犁等地区相接壤,较近的地理距离以及野兔较强的迁移能力可能为北部和西北部组群间的基因流动提供了条件。
系统发育树中,Clade B是来自中部和东部组群的野兔,在形态分类学上属于托氏兔中亚亚种。Fst值和基因流分析显示(表4),西域亚种和中亚亚种间(北部/西北部与中部或东部)的Fst值(0.306~0.376,P<0.01)比亚种内(北部与西北部或中部与东部)的Fst值(0.101~0.279,P<0.05)高,同样地,亚种间的基因流比亚种内的基因流小,说明新疆托氏兔亚种间分化程度更高、基因流更有限。AMOVA分析结果(表3)也证实了这一点,当按照两个亚种分组时,亚种间的变异(19.400,P<0.01)大于亚种内的变异(13.070,P<0.01)。值得注意的是,中部和东部组群的样本在系统发育树中(图2)聚在一大枝上,然而中介网络图上(图3)从一大枝分成两个小枝,而且中部和东部组群间基因流较小,为0.646,Fst值(为0.279,P<0.01)较大,说明存在很大的遗传分化[30]。但是,当按照托氏兔西域亚种、中部和东部组群分组进行AMOVA分析时(表3),结果显示种群内的变异贡献率最大,为67.560(P<0.01),组群间的变异不显着。结合地理分布来看,虽然中部与哈密地区相接壤,但是达坂城和托克逊与哈密地区中间有广阔的吐鲁番盆地和哈密盆地,地理距离相对较远,可能阻碍了中部与东部组群间的基因交流,促进了分化。
3.3 新疆托氏兔的种群历史动态
对于种群历史动态的推断,常常通过中性检验并结合错配分布进行分析。当中性检验(Tajima`D和Fu’sFs)的值为负值(P<0.05),且错配分布呈单峰分布时,说明所研究的群体可能在历史上经历过种群扩张;而群体大小保持稳定时,中性检验不显着,错配分布曲线则呈现多峰曲线分布[31]。本研究的中性检验结果显示(表2),新疆托氏兔和各组群的Tajima`D值均不显着,并且核苷酸错配分布呈现多峰(图4A);而本研究中新疆托氏兔的Fu`sFs为负值(-15.100)且差异极显着。据报道,物种具有较大的Fu`sFs值、且为负值时,说明该物种在近期积累了较多变异,并且近期可能经历过群体扩张[32-33]。此外,EBSPs分析结果显示新疆托氏兔在距今大约0.01 Ma(百万年)时发生过种群扩张(图4B),而文献表明中全新世(8~4 Ka)[34]时期气候适宜、植被覆盖增加,这些因素可能为托氏兔的扩张提供了有利条件[35-36]。综合来看,本研究初步推测新疆托氏兔在距今大约0.01 Ma时经历过近期的种群扩张,后续应对托氏兔的种群历史进行全基因组层面更深入的研究。
4 结 论
综上所述,本研究基于mtDNA的CO1和D-LOOP基因的综合分析表明,新疆托氏兔单倍型丰富,单倍型多样性和核苷酸多样性均较高,具有较强的应对环境变化的能力。新疆托氏兔的分布具有较明显的系统地理分布格局,且不同组群间存在一定程度的遗传分化和基因交流。新疆托氏兔亚种间分化较大,基因流较小,西域亚种的北部和西北部组群间存在较强的基因流和较小的遗传分化,但中亚亚种的中部和东部组群间出现了较大的分化,可能是由于较远的地理距离所致。此外,初步推测新疆托氏兔近期经历过种群扩张。



