邓敏儿,李 娜,郭亚琼,冯耀宇,肖立华
(华南农业大学,岭南现代农业科学与技术广东省实验室,广州 510642)
原生动物包含许多人兽共患和重大动物疫病寄生虫,严重影响人类和动物健康。由于生活史复杂,不少寄生虫缺乏完成整个生活史的体外培养技术,原虫的功能基因组学和分子生物学研究因此受阻。1993年,弓形虫[1-2]和疟原虫[3]被报道可以利用同源重组实现报告基因的表达,此后其他原虫的反向遗传学工作平台也逐步建立。最近20年,原虫的反向遗传学研究取得长足进展。通过设置同源臂的方法,同源重组技术为原虫的基因定位、功能研究、抗原筛选等研究提供有力支持[4]。但同源重组的基因编辑系统存在转染效率较低、需要的同源臂较长、筛选过程时间长的问题。有的原虫(如隐孢子虫)基因组小,缺乏同源重组修复系统,较难开发同源重组基因编辑体系。
CRISPR(成簇的规则间隔的短回文重复序列)是在细菌和古细菌中发现的一种天然存在的适应性免疫系统,其作用是破坏噬菌体和质粒等外源DNA序列[5-7]。最早研究的源自化脓链球菌的Cas9蛋白(SpCas9,最初被命名为CSN1)是目前最常用的Cas9蛋白。此外,2017年报道源自金黄色葡萄球菌的SaCas9蛋白在克氏锥虫基因编辑中达到几乎100%的基因敲除效率。2012年2个实验室的工作证明优化后的CRISPR/Cas9系统可用于生物的基因编辑,并将该系统工作所需的2条RNA进一步简化为一条向导RNA(gRNA),实现人工设计靶向不同基因区域的gRNA和Cas9蛋白,完成定向高效的DNA双链切割[6-7]。CRISPR/Cas9系统被《Science》杂志评选为2013年度最重要的科学突破之一,自应用以来在生命科学领域大放异彩,在寄生虫研究上也从疟原虫和弓形虫,逐步推广到在利什曼原虫、隐孢子虫和艾美耳球虫等的应用。CRISPR/Cas9系统在人工设计的前提下,完成定向DNA双链断裂(DSB),供体的模板质粒只需更短的同源臂,即可完成准确高效的基因编辑。该系统还可针对多基因家族[8],利用少量的gRNA完成高通量全基因组筛查工作,是寄生虫的基因功能研究强有力的工具。本文将从CRISPR/Cas9系统的工作原理和目前在寄生原虫中的主要应用进行介绍。
1 CRISPR/Cas9系统工作原理
在CRISPR/Cas9系统出现之前,原虫的基因编辑主要通过同源重组的方式实现。将带有一定长度(一般至少400 bp)同源臂的目的基因的质粒转入寄生虫,利用同源重组让目的基因插入原虫基因组同源臂中,从而实现基因编辑[9]。该方法所需时间长,转染效率低(双质粒共转染的效率更低),药物筛选耗时较长,一般只能进行单基因的研究。
CRISPR/Cas9系统分为I型、II型和III型,其中III型在基因编辑上应用最广泛——主要通过Cas9蛋白对目标DNA双链切割。2013年,CRISPR/Cas9系统被简化为只需要Cas9蛋白和20 bp的gRNA即可完成定向DNA编辑[6-7]。CRISPR/Cas9系统工作原理及修复机制如图1所示,其中DNA双链断裂修复主要通过3个机制:1)同源直接修复(homologous direct repair,HDR)。当存在具有断裂位点上下游同源臂的DNA时,以此为模板进行修复。2)非同源末端连接(non-homologous end joining,NHEJ)。当缺少供体模板时,在两个断口直接连接。3)微同源末端连接(microhomology-mediated end joining,MMEJ)。利用微同源小片段切除非同源的单链DNA,断口处直接连接。
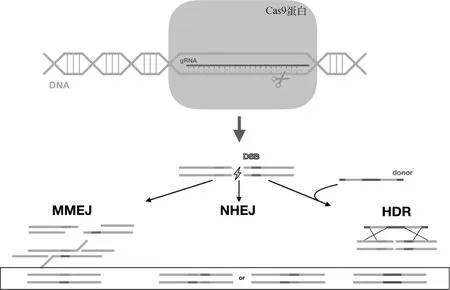
图1 CRISPR/Cas9系统工作原理及修复机制Fig.1 Principle and repair mechanism of the CRISPR/Cas9 system
除常用的电转含有Cas9和gRNA质粒的方法外,还可以将Cas9蛋白和gRNA复合物直接转入寄生虫体内。后者在克氏锥虫中达到转染效率接近100%的效果[10]。CRISPR/Cas9系统显着提高原虫的转染效率,并且可通过建立gRNA文库的方式,完成双基因、甚至多基因的研究,为多基因家族功能研究提供高通量快速筛查的高效准确工具。
2 CRISPR/Cas9系统在原虫中的应用
寄生原虫基于CRISPR/Cas9的基因编辑技术开展较早,可追溯到20世纪80年代末、90年代初[1-3, 11-16]。2014年Shen等[17]和Zhang等[18]分别将CRISPR/Cas9技术引入弓形虫和克氏锥虫的反向遗传学研究,自此CRISPR/Cas9系统逐渐在研究原虫的基因定位、基因敲除、基因家族高通量筛选等方面崭露头角[19-60](部分原虫基因编辑技术发展情况见图2)。与传统的同源重组技术相比,CRISPR/Cas9系统不仅具有转染效率更高、周期更短、准确性更高等优点,并且可在较短的时间内对同源性高的基因家族进行高通量筛选。此外,CRISPR/Cas9系统可结合多种反向遗传学研究策略,如利用CRISPR/Cas9系统标记关键基因[19]、结合类植物生长素诱导条件性降解系统(auxin-inducible degron,AID)[20]研究必需基因的功能和表型、利用改良的CRISPR/dCas9系统进行表观遗传学研究[21-22]等,显着扩展其应用领域,为假定基因的功能研究提供高效工具。
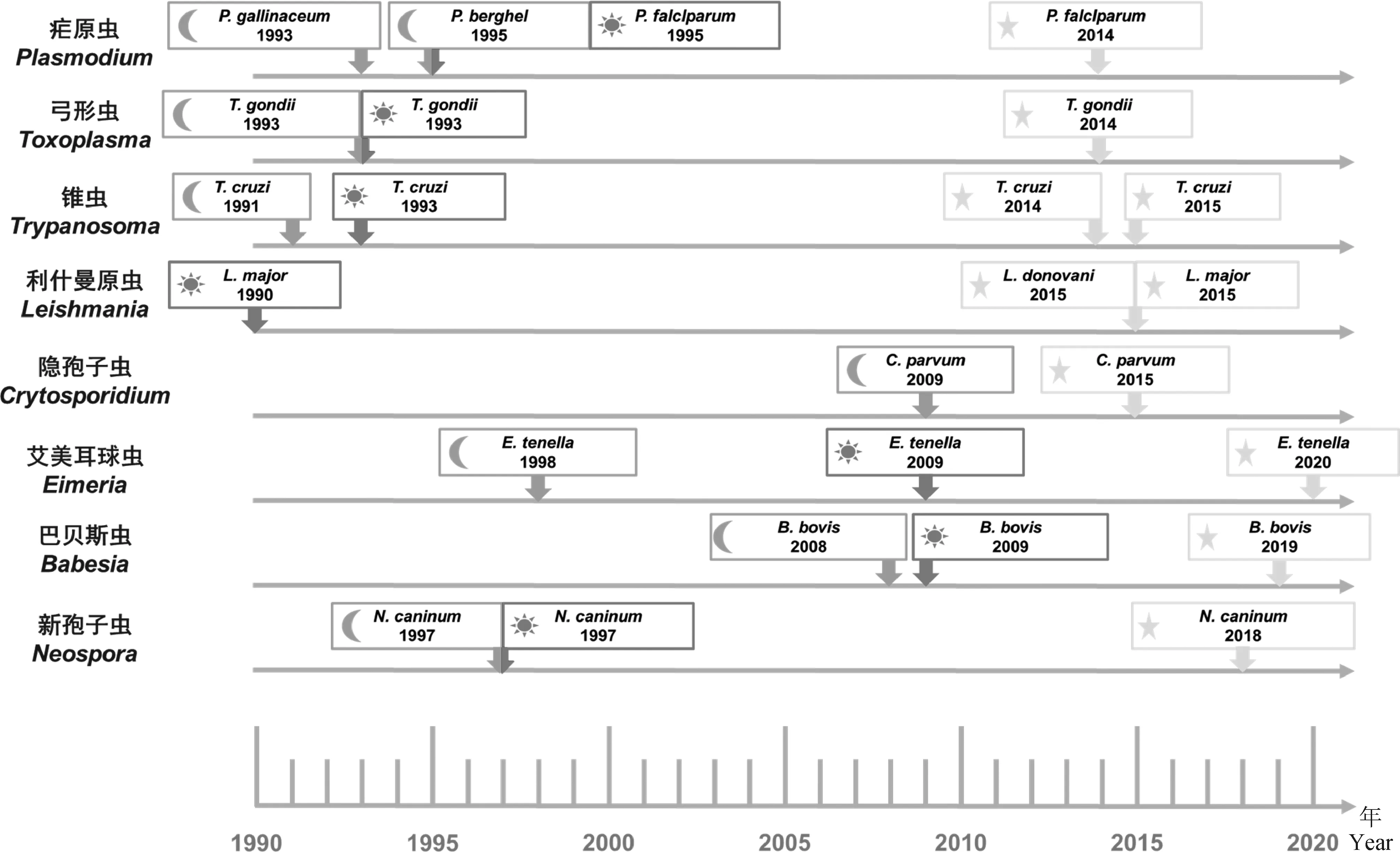
月亮、太阳和星星标记分别代表瞬时转染、稳定转染和CRISPR/Cas9技术[1-3,9,11-13,15-19,23-24,49-63]Moon, sun, and star shapes indicate transient, stable transfection, and CRISPR/Cas9-based editing, respectively[1-3,9,11-13,15-19,23-24,49-63]图2 部分原虫基因编辑技术发展时间线Fig.2 Timeline of the development of genetic manipulation methods in some parasitic protozoa
2.1 疟原虫
2014年,2个研究团队分别报道了CRISPR/Cas9系统在疟原虫上的应用。Wagner等[23]利用T7 RNA聚合酶产生的sgRNA和Cas9蛋白对恶性疟原虫的基因组进行编辑,分别靶向富含组氨酸的疣状突起相关蛋白基因(kahrp)和红细胞结合抗原175基因(eba-175),证实该系统在恶性疟原虫上具有较高的基因编辑效率(50%~100%)。恶性疟原虫缺乏NHEJ途径,CRISPR/Cas9系统可显着提高疟原虫的基因编辑效率。
同年,Ghorbal等[24]同样构建对恶性疟原虫基因编辑的CRISPR/Cas9系统,利用Cas9质粒及含sgRNA和人二氢叶酸还原酶药筛基因(hdhfr)的供体模板质粒共转染的方案,敲除表达绿荧光的NF54株的egfp基因。此时Cas9蛋白为瞬时表达。3周内得到的WR耐药虫株PCR和荧光检测均证实egfp基因被成功敲除。供体模板质粒线性化后转染,证实与环形质粒几乎一致的转染效率,而且具有转染4 d后线性化模板自动丢失的优点,避免供体模板对整合效果检测的影响。除了靶向egfp基因,同样的方案应用在内源性的非必需基因kahrp上得到了相似的效果。后续研究利用该系统对orc1基因完成不带标签的定点突变,发现基因敲除的克隆株具有青蒿素抗性。
CRISPR/Cas9系统作为强大的基因编辑工具,为疟原虫的基因编辑研究提供有力支持。如2015年Nacer等[25]利用该系统鉴定PfVAP1(毒力相关蛋白1)是恶性疟原虫细胞黏附的关键因素。Mogollon等[26]2016年改良Ghorbal等团队的研究方法,重新设计sgRNA和供体质粒,证实PfVAP1在FCR3疟原虫的黏附过程中的作用。Bryant等[27]在2017年使用该系统完成恶性疟原虫var2csa基因编辑,发现缺失内含子不影响该基因对正常调控。2019年Xiao等[22]应用改良的 CRISPR/dCas9 系统对寄生虫入侵相关基因的转录进行表观遗传调控,并证明dCas9可以将融合的表观遗传调节因子[如组蛋白乙酰转移酶(HAT)或去乙酰化酶(HDAC)结构域],完成恶性疟原虫基因过表达或转录敲低。同年Mohring等[28]实现CRISPR/Cas9系统在诺氏疟原虫基因组编辑上的应用,但存在同源重组修复效率低的问题。2021年Boltryk等[29]报道利用CRISPR/Cas9建立NF54诱导的配子体产生虫株,实现大规模同步产生定向分化的配子体,为疟疾传播阶段研究提供宝贵工具。
2.2 弓形虫
弓形虫具有较为成熟且容易的体外培养和遗传研究系统,突变诱导、基因标记、基因敲除和基因替换等较其他原虫容易[8]。同时弓形虫具有活跃的NHEJ途径,DNA随机整合效率高,因此降低同源重组的精确性。
Shen等[17]早在2014年对CRISPR/Cas9系统进行改造,将含有靶向尿嘧啶磷酸核糖转移酶(UPRT)基因的sgRNA和Cas9-NLS-GFP的质粒电转入I型RH株中,有20%~30%的弓形虫为Cas9-GFP阳性。UPRT失活可产生氟脱氧核糖(FUDR)抗性,故FUDR耐药性的噬斑试验证实约有10%的Cas9-GFP阳性弓形虫UPRT失活。FUDR耐药的单克隆虫株目标区域的基因测序结果证实UPRT存在短缺失或插入突变。重复上述转染试验时共转包含乙胺嘧啶耐药基因DHFR的供体质粒,同时用乙胺嘧啶筛选,观察到70%~100%的乙胺嘧啶抗性寄生虫同样抗FUDR。乙胺嘧啶抗性虫株的单克隆的PCR结果证实超过45%的虫株为DHFR的正确整合,说明该系统能有效增强gRNA靶向区域的局部重组,提高基因敲除频率。改变供体质粒后重复试验,成功删除UPRT基因,说明该系统可同样应用于较大基因区域的敲除。为研究该系统是否与同源重组的基因编辑相同、存在需要相对较长同源臂的局限性,该团队将长同源区域(750~900 bp)、20 bp侧翼区域和缺乏同源性的dhfr的靶向效率做比较,发现三者均显示高于50%的正确整合效率,表明该系统可用于提高特定位点的非同源整合效率。该团队用靶向丝氨酸苏氨酸激酶(ROP18)的质粒对I型的GT1株的敲除及敲除虫株的回补试验,证实该系统可广泛地应用于弓形虫基因编辑。2017年该团队优化CRISPR/Cas9系统,通过引入突变降低Cas9蛋白毒性,并提供弓形虫CRISPR/Cas9系统应用操作指南[30]。
Sibley团队在2017年报道2篇CRISPR/Cas9系统和类植物生长素诱导的降解因子(AID)系统在弓形虫基因功能研究中的应用:Long等[31]检测类钙调蛋白(CaM)的功能,证实CaM与肌凝蛋白(MyoH)共定位,且与弓形虫入侵密切相关;Brown等[32]检测蛋白激酶(PKGs)的功能,证实PKG I为弓形虫生长必需蛋白,而PKG I存在的前提下,PKG II为弓形虫的非必需蛋白。
Sidik等[33]在2014年构建包含靶向速殖子表面蛋白基因(sag1)的嵌合体RNA(chiRNA)和带flag标签带Cas9蛋白的质粒。在未经筛选的情况下,RH株的sag1敲除效率约为20%。用缺少NHEJ修复机制的ΔKU80株重复上述试验,寄生虫数量与RH株相比减少10倍,而转染效率相当。根据转染替换Cas9成dhfr的相似质粒的2种虫株的噬斑试验对比,进一步证明弓形虫在DNA双链断裂后依赖NHEJ修复。该研究还利用较短的同源臂(40 bp)定点插入模板DNA,实现对内源性基因cdpk3的标记。该团队在2016年设计gRNA文库,对弓形虫基因组进行高通量筛选[8]。
Cas9蛋白在弓形虫中存在内在毒性,传代过程中该基因频繁丢失,Markus等[34]在2019年发现优化表达载体和gRNA的设计能显着降低Cas9蛋白的毒性,认为gRNA同时具有基因组靶向、促进Cas9蛋白的整合和降低Cas9毒性、提高适应性的功能。
CRISPR/Cas9系统在弓形虫研究中应用以来,已成为弓形虫高通量筛选[8,35-37]、基因功能验证[38-39]、致病机制[40-41]、免疫互作[35, 42-44]、代谢分析[45]、药物机制[36, 46]、疫苗开发[47]等研究必不可少的工具,并在此基础上开发的AID系统[20]、转录抑制(基因条件性敲低)[48]等新工具,为寄生虫研究提供更广阔的研究前景。
2.3 艾美耳球虫
Tang等[57]报道CRISPR/Cas9系统在柔嫩艾美耳球虫高效基因编辑的应用。首先通过瞬时转染His4启动子介导的含有黄荧光基因(yfp)标记的Cas9质粒进入子孢子,体外培养24 h观察到球虫细胞核发黄荧光,证实His启动子介导的Cas9基因可在细胞核内表达。然后构建靶向黄荧光基因的sgRNA的质粒转染已有同时发黄荧光和红荧光的EtER虫株,在CRISPR/Cas9系统作用下,转染成功的球虫内因为黄荧光基因发生断裂而失去黄荧光。经过4次传代和流式细胞术筛选,发现黄荧光阳性比例减少到89.5%,说明缺少供体质粒的情况下,球虫偏向于利用NFEJ途径进行自我修复。另外还构建靶向mic2基因末端的sgRNA质粒和带有红荧光基因和药筛基因的供体质粒,对野生型球虫子孢子共转染,并利用PCR和IFA进行验证。试验证明CRISPR/Cas9系统可用于球虫内源性基因和外源性基因的基因编辑,但效率远低于弓形虫或疟原虫,推测可能是双质粒共转染或转染效率低所致。
Hu等[19]利用同源重组技术成功构建在生活史各阶段均能稳定表达Cas9蛋白的柔嫩艾美耳球虫株。该虫株转染不同sgRNA质粒,能进行高效的球虫单基因编辑。向该虫株转染含有靶向外源的黄荧光基因的sgRNA的质粒,证实该系统切割效率可达29%。接着转染针对内源性蛋白EtGRA9的sgRNA和供体质粒,为EtGRA9加上荧光标记,在共聚焦显微镜下发现EtGRA9标记信号存在于囊泡中而不是致密颗粒中,说明EtGRA9属于分泌蛋白。同时设计靶向AP2家族33个基因的sgRNA,经过6次转染试验,发现只有10个AP2家族的基因被靶向破坏,其中可检测9个gRNA(另一个有效读数较低,表明破坏该基因引起虫体生长缺陷)。该试验证明球虫AP2家族中,23个基因在球虫生长繁殖必不可少,为球虫AP2家族研究奠定基础,也证明该工具虫株可以对球虫进行高效的单基因编辑。
2021年Cheng等[64]报道FnCas12a/crRNA和RNP技术可用于柔嫩艾美耳球虫的基因编辑。该团队成功靶向组蛋白H4基因导致其DNA双链断裂,另外还利用eYFP标记内源性EtActin蛋白,观察不同时期肌动蛋白的定位情况。同年Mohsin等[65]报道体外试验中Cas9 mRNA与sgRNA结合可降低卵囊的孢子化率和存活率,为抗球虫试剂研发提供新思路。
2.4 锥虫
克氏锥虫由于缺少RNAi的机制,故在功能遗传学研究中受到限制。因此,克氏锥虫的研究需要更强大的反向遗传学研究工具。
2014年Peng等[49]报道CRISPR/Cas9系统在克氏锥虫的首次应用。构建稳定表达绿荧光(GFP)和Cas9(源自化脓链球菌)的虫株后,再分别转入3种靶向gfp的sgRNA。在没有模板的情况下,克氏锥虫的Cas9诱导的DSB的修复缺乏NHEJ途径,只能通过MMEJ进行,产生各种大小的基因缺失。在提供模板tomato标记的情况下,CRISPR/Cas9的基因编辑效率比仅用模板质粒的同源重组方式更高。除外源性基因gfp的破坏外,还利用CRISPR/Cas9分别成功敲除内源性的α-微管蛋白基因、组氨酸裂合酶(HAL)基因和脂肪酸转运蛋白(FATP)基因。此外,还利用CRISPR/Cas9系统对同源性高的多基因家族进行研究,仅设计3个sgRNA即可靶向β-半呋喃糖基糖基转移酶(GAL)家族的65个基因,3次转染后,63%的gal基因获得至少1个靶位点突变,显着敲低该酶基因家族的表达,且未观察到脱靶现象。最后,该研究利用靶向Cas9的sgRNA对Cas9基因破坏,挽救表达Cas9的虫株生长缺陷。上述研究证明CRISPR/Cas9系统可快速高效敲除单拷贝或多拷贝基因,但存在虫株生长速度慢和Cas9活性随时间降低的问题。
Lander等[66]在2015年报道CRISPR/Cas9编辑系统对鞭毛杆蛋白1和2敲除研究,试验结果亦反映出该系统存在药物筛选时间长(数周)和无法研究必需基因的问题。
Soares等[10]在2017年采取源自金黄色葡萄球菌(SaCas9)的Cas9蛋白和gRNA复合物电转的方式,建立接近100%转染效率的克氏锥虫快速基因编辑的系统。首先利用SaCas9/eGFP sgRNA复合物对表达绿色荧光的克氏锥虫进行转染,7 d即达到超过97%寄生虫的敲除,且对照组gfp自发损失极小。其中基因敲除效率与gRNA选择、RNP复合物浓度及电转次数有关。接着构建具有SaCas9-egfp/gRNA复合物,该复合物大小与SpCas9大致相同且具有Cas9活性,用SaCas9-egfp复合物转染低于7%,远低于SaCas9/gRNA复合物转染效率(大于75%),证实转染SgCas9/gRNA复合物无法进行基因敲除与Cas9-RNP较大有关。通过分别转染不同品系和生活史阶段的锥虫,证实SaCas9/gRNA复合物可转染锥鞭毛体,转染不同表型和基因型的虫株效率相当。单次转染靶向2个基因的RNP复合物,单敲除效率可高达90%,双敲除效率为55%。利用含有终止密码子的修复模板和RNP,可以使galf这类克氏锥虫必需单基因敲除时完成单等位基因敲除。利用RNP和修复模板分别对NTR和CYP进行敲除,再用对应的抗性药物处理,发现RNP介导的必须基因敲低可应用于寄生虫药物基因的研究。此外,还可以用RNP和携带表达标签(如HA)的修复模板对内源性基因标记,在标记的抗体染色和流式细胞术的配合下不需药物筛选,快速高效完成内源基因标记。SacCas9/sgRNA复合物可同样适用于布氏锥虫和利什曼原虫的研究。同年,Beneke等[67]报道建立稳定表达Cas9蛋白的布氏锥虫虫株,结合PCR扩增产物可用于基因高效快速的编辑。该技术亦可应用于利什曼原虫反向遗传学研究:该团队利用CRISPR/Cas9建立鞭毛突变体文库[68],研究利什曼原虫鞭毛蛋白组功能和真核生物的鞭毛运动。
CRISPR/Cas9基因编辑技术在克氏锥虫上成功应用,极大加快锥虫反向遗传学研究进展。1993年用传统基因编辑技术成功敲除克氏锥虫[12],此后20年仅20篇关于克氏锥虫的基因编辑报道。自CRISPR/Cas9基因编辑技术成功应用后至今,克氏锥虫完成基因编辑的基因超过67个[69]。
2.5 利什曼原虫
Zhang等[18]在2015年利用杜氏利什曼原虫的rRNA启动子和肝炎三角洲病毒(HDV)核酶构建gRNA表达载体,与Cas9蛋白转入杜氏利什曼原虫中,靶向米替福新(MLF)转运蛋白(LdMT)。LdMT的两个等位基因均突变时,寄生虫表现为MLF耐药。从MLF耐药率可以看出,含有HDV核酶的gRNA质粒让gRNA质粒转染效率从0.5%增至12%。对转染后表现MLF耐药的利什曼原虫进行测序,发现利什曼原虫以MMEJ而不是NHEJ途径修复DSB。对比转染含有终止密码子的模板质粒,发现转染效率提高2~6倍,说明HDR途径可以显着改善定点插入的基因编辑能力。用该系统将ble基因定点替换Ldbpk_241510.1基因(一种多药抗性蛋白样基因),博来霉素药物筛选的阳性虫株的PCR结果证明该系统可以定点插入外源蛋白和敲除非必需基因。用同样的方法靶向必需基因TOR1基因时,发现转染效率较低,并只能破坏其中一个等位基因,说明该系统能部分敲除利什曼原虫的必需基因。还利用该系统成功在LdMT蛋白的基因位点加上gfp标签,以精确标记内源性基因。最后,该研究利用锤头核酶和HDV核酶构建效率更高的双gRNA表达载体,提高利什曼原虫基因编辑效率。2017年该团队优化原有基因编辑技术,构建Cas蛋白和gRNA共表达载体,成功对杜氏利什曼原虫、硕大利什曼原虫及墨西哥利什曼原虫完成基因编辑,并且利用多靶向gRNA实现对杜氏利什曼原虫AP2家族的11个基因敲除[70]。
Sollelis等[51]在2015年构建稳定表达Cas9的硕大利什曼原虫株(命名为M756株),再向M756株转入同时包含靶向鞭毛杆2基因pfr2(位于鞭毛杆,属于非必需基因)的gRNA和作为供体的DHFR-Ts药筛基因。PCR和FISH检测结果表明,转染后的M90株属于阳性和阴性虫株混合群体。从M90株筛选的8个克隆种,2个克隆被鉴定为成功转染的虫株。为分析CRISPR/Cas9系统的脱靶效应,接着对2个阳性克隆株进行全基因组测序,软件评估出的排名前十的潜在脱靶位点的20 nt内均未发现插入缺失标记,证明未出现脱靶现象。
虽然RNAi技术在利什曼原虫中已有广泛应用,但CRISPR/Cas9系统亦可作为与之互补的基因编辑工具,二者为不同目的和应用的研究提供重要技术支持。2019年Shrivastava等[71]利用CRISPR/Cas9系统建立LeishIF4E-3(+/-) 突变体,该突变体对巨噬细胞的感染性严重降低,且形态发生改变。2021年Baker等[72]用 mNeonGreen 荧光蛋白标记蛋白激酶以进行定位研究,同时建立ePK以及PIKK两种蛋白激酶突变虫株,评价蛋白激酶调节寄生虫的复制、分化等的作用。
2.6 隐孢子虫
Vinayak等[53]在2015年报道CRISPR/Cas9系统在微小隐孢子虫的应用,该团队在2017年提供隐孢子虫转基因技术的详细试验方法[73]。首先在感染HCT-8细胞体外培养的条件下,利用同源重组将荧光素酶基因(nluc)插入烯醇酶基因(一种持家基因)后,利用荧光强度作为寄生虫计数指标。向野生虫株转染nluc缺陷质粒(在nluc基因中插入终止密码子),在转染靶向nluc缺陷片段的gRNA和一小段DNA双链模板后表达荧光,证实CRISPR/Cas9系统可应用在微小隐孢子虫的基因编辑研究。为提高转染效率,选择氨基糖苷磷酸转移酶NEO(对巴龙霉素有抗药性)作为药筛基因,通过Nluc-Neo融合表达的方式,在巴龙霉素作用下筛选转染的寄生虫。通过对比试验可以发现,同时共转Nluc-Neo和Cas9质粒的寄生虫排卵囊量迅速恢复到与无药野生型组小鼠相似水平,而只转染Nluc-Neo或nluc的小鼠逐步被巴龙霉素治愈,这说明CRISPR/Cas9系统基因编辑效率高于同源重组。对小鼠传代后的Nluc-Neo的寄生虫分别加巴龙霉素进行小鼠体内培养和细胞体外培养,小鼠传代与野生型虫株排卵囊量对比,可明显看出Nluc-Neo虫株对巴龙霉素耐药,免疫印迹、IFA和荧光素试验均证实转染虫株是稳定的转基因虫株。同时,因为微小隐孢子虫具有两条合成dTMP的途径,其中一条为TK酶利用EdU合成dTMP。因此设计针对tk基因的gRNA和Nluc-Neo作为报告基因的供体模板,对隐孢子虫的内源性基因tk完成敲除。该报道中还优化电转程序和缓冲液体系,达到提高10倍电转效率的效果。此外,该团队根据基因组测序结果认为隐孢子虫缺少非同源末端连接的DNA修复途径,在使用CRISPR/Cas9系统时,必需要提供供体DNA。2020年该团队在双环氮杂环丁烷治疗隐孢子虫病的研究中,利用CRISPR/Cas9系统在隐孢子虫中插入苯丙氨酰-tRNA合成酶基因(pheRS基因)并使之产生药物抗性,证实苯丙氨酰-tRNA 合成酶被鉴定为双环氮杂环丁烷的靶标[74]。同年,Choudhary等[75]报道基于大肠杆菌二氢叶酸还原酶降解结构域 (DDD) 和甲氧苄啶 (TMP)和 CRISPR/Cas9系统建立第一个隐孢子虫条件性敲降技术,并完成cdpk1基因进行条件性敲除,有助于隐孢子虫的必需基因功能研究。
2021年Xu等[76]在INS1蛋白功能研究中使用CRISPR/Cas9系统标记或敲除ins1基因,发现INS1蛋白参与大配子体形成或成熟。同年,Hasan等[77]利用CRISPR/Cas9系统对隐孢子虫耐药性进行研究。该团队证实Cp MetRS合成酶发生突变后隐孢子虫能正常生长并对MetRS抑制剂2093药物产生抗性。
2.7 其他原虫
CRISPR/Cas9系统在新孢子虫(Neosporacaninum)的应用首次报道出现在2018年。Arranz-Solís等[63]首次在犬新孢子虫利用CRISPR/Cas9系统实现基因编辑,另外Nishikawa等[78]在同年也报道应用CRISPR/Cas9系统进行犬新孢子虫NcGRA7蛋白的敲除及功能鉴定。前者报道中利用原本用于弓形虫的质粒系统,成功应用于新孢子虫研究。该研究一方面设计2个针对Nc-1-GFP表达虫株中的GFP蛋白的gRNA,实现外源基因的敲除;另一方面针对新孢子虫的gra7基因设计2个gRNA及相应的供体质粒,完成内源基因gra7的敲除。此外,该团队证实CRISPR/Cas9系统可应用于犬新孢子虫的基因编辑。该团队在2022年报道弓形虫的生长素诱导条件性降解系统(AID)可在新孢子虫中成功应用[79]。
2019年Hakimi等[60]利用单个表达 Cas9、gRNA 和供体模板DNA的质粒,成功用于牛巴贝斯虫(Babesiabovis)的基因编辑。该团队用单质粒系统对牛巴贝斯虫SBP3蛋白末端融合表达myc标签实现蛋白定位,并在感染巴贝斯虫的红细胞表面呈现斑块状染色。该报道还设计针对细胞质抗氧化酶过氧还蛋白基因tpx-1实现单质粒的点突变,将49位的过氧化半胱氨酸突变为丝氨酸。另外,该团队还构建敲除tpx-1基因并替换为gfp基因的虫株。通过tpx-1的突变株和敲除株与原有的野生型的对比发现,前两者均表现出对药物硝普钠(SNP)敏感的特性。
3 展 望
CRISPR/Cas9系统可以对单基因甚至多基因进行高效、精确和快速的编辑,但在寄生原虫基因编辑的应用上仍存在部分问题。第一,CRISPR/Cas9系统应用时可能存在脱靶效应,应进一步完善gRNA的设计方法,在试验过程中注意对潜在脱靶位点的检查。第二,Cas9蛋白较大,整合到寄生虫基因组中较为困难,对于寄生虫稳定表达Cas9蛋白带来挑战。第三,Cas9蛋白对寄生虫有一定毒性,如克氏锥虫的Cas9蛋白表达株存在生长缺陷[49],弓形虫表达的Cas9蛋白自发丢失[34]。可在表达Cas9蛋白的同时加入“陷阱sgRNA”的方式降低毒性[34],或对Cas9蛋白优化,选择更高效安全的Cas9蛋白(如金黄色葡萄球菌的Cas9蛋白[10])。第四,与疟原虫和弓形虫相比,隐孢子虫、球虫等部分原虫可用转基因虫株筛选的抗性基因较少,限制转染效率的提高及基因编辑策略的发展。
CRISPR/Cas9系统在基因编辑应用的不到十年里中,已经在生命科学领域占有重要地位。除了高效的单基因编辑、精确的点突变外,在弓形虫等原虫中出现利用不同策略实现多基因家族编辑、全基因组编辑筛选的报道,展示CRISPR/Cas9系统功能的多样性。虽然仍存在上述的问题,但相信通过科研人员的努力,将来CRISPR/Cas9系统可作为高效便捷的遗传操作工具,可应用在多种寄生性原虫的基因家族功能研究、全基因组功能分析、数量性状研究、药物筛选与耐药基因研究、表观遗传学调控等深入研究之中,并具有应用在往日较难开展反向遗传学操作的寄生性原虫中的潜力,可以为原生动物的反向遗传学、基因功能和疫苗开发等研究提供强有力的技术支持,有望在寄生性原虫的基础研究与防控开展上大放异彩。



