文 强,唐明美,陈 玲,彭章丽,何月娟,唐正洪
(1.遵义医学院附属医院 呼吸二科,贵州 遵义 563099;2.山东省临沂市人民医院东医疗区 内三科,山东 临沂 276000)
临床医学研究
结核分枝杆菌包涵体蛋白Rv3480c的克隆、表达与纯化
文 强1,2,唐明美1,陈 玲1,彭章丽1,何月娟1,唐正洪1
(1.遵义医学院附属医院 呼吸二科,贵州 遵义 563099;2.山东省临沂市人民医院东医疗区 内三科,山东 临沂 276000)
目的应用分子生物学技术对结核分枝杆菌重组抗原Rv3480c基因进行克隆、表达和纯化。方法以结核分枝杆菌标准菌株H37Rv基因组为模板,通过聚合酶链式反应(PCR)扩增,克隆Rv3480c基因片段,产物经验证后与载体质粒pET28a连接重组质粒,将验证构建成功的重组质粒转化入表达宿主E.coli BL21(DE3)菌体内,经异丙基半乳糖苷(IPTG)诱导蛋白表达,溶解包涵体,并通过多次低温冻融、不同浓度尿素复性方法进行包涵体蛋白质的纯化。结果扩增后基因电泳试验及测序证实成功重组目的基因,用尿素溶液和磷酸盐缓冲液(PBS)透析和纯化重组蛋白,Western-Blot证实成功重组结核分枝杆菌抗原Rv3480c。结论应用分子生物学技术成功获得结核分枝杆菌Rv3480c蛋白,为其结构和功能的进一步研究奠定了基础,并为包涵体蛋白的纯化提供了新的技术思路。
结核分枝杆菌;Rv3480c;包涵体;克隆;纯化
结核病是由结核分枝杆菌感染引起的慢性传染病,是全球范围内单一病原体引起人类死亡人数最多的疾病之一[1]。2015年,全球估计有1040万新发结核病患者,结核病已成为导致20~59岁女性死亡的五大重要原因之一,也是威胁儿童健康的重要疾病,结核病更是HIV阳性感染者死亡的主要杀手,中国是全球结核病患者比例居前六位的国家之一[2]。
面对如此严峻的结核病疫情,快速诊断和早期治疗是控制结核病疫情的重要手段[3]。卡介菌纯蛋白衍化物(PPD)皮试、结核菌涂片/培养检查、γ干扰素释放试验/TSPOT-TB等技术方法是目前结核病实验室诊断的重要手段。在这些常用的诊断方法中,PPD敏感性较高但特异性差,细菌学检查是诊断结核病的金标准,但抗酸杆菌涂片阳性率低,而传统结核分枝杆菌培养周期长,延误结核病早期诊治,γ干扰素释放试验/TSPOT-TB敏感性和特异性均高,但不能区分活动性结核病和潜伏性结核感染,且价格昂贵[4]。同时,传统的实验室诊断技术不能满足肺外结核病的临床应用[5],因此,我们急需要寻找一种快速、灵敏、特异性高且经济的检测方法。
结核分枝杆菌感染人体后在机体内激发免疫应答,产生针对结核菌菌体蛋白的特异性抗体。血清学诊断通过结核分枝杆菌特异性抗原检测患者血清中特异性抗体水平诊断结核病。目前国内外已有多种结核分枝杆菌特异性抗原作为被检靶标,例如ESAT6[6]、M.tb81、M.tb8、M.tb48、DPEP (MPT32)等[7],其灵敏度和特异度各研究报道不尽一致,仍需继续筛选与结核相关的高特异性抗原,开发更有诊断价值的血清学诊断方法。Rv3480c蛋白是本课题组利用蛋白芯片技术筛选出的特异性抗原之一,是结核菌分泌到菌体外的蛋白抗原,其结构和功能尚未清楚,我们选取该蛋白进行克隆、表达和纯化,为进一步的功能验证、结构分析以及血清学诊断试剂盒的制作奠定基础。
1 材料与方法
1.1 材料 人型结核分枝杆菌标准菌株H37Rv由贵州省疾病预防控制中心馈赠。原核表达载体pET-28a购于Novagen公司。DH5α和BL21(DE3)感受态细胞购自天根生化科技有限公司。EcoR I限制性内切酶、Hind III限制性内切酶及T4连接酶均购于TaKaRa公司。Pfu高保真PCR反应试剂盒购于TransGen Biotech。质粒小提试剂盒、琼脂糖凝胶回收试剂盒、500bp DNA marker购于天根生化科技有限公司。蛋白预染marker购自Thermo公司。组氨酸(6x-His)抗体购自美国Sigma公司,二抗购自美国Abcm公司。
1.2 主要仪器 超净工作台购于中国上海智诚分析仪器公司。超声细胞粉碎仪购于中国宁波新芝生物科技公司。低温高速离心机购于德国Eppendorf公司。半干转电泳仪购于美国Bio-Rad公司。恒压恒流电泳仪购于美国Bio-Rad公司。凝胶成像系统购于美国Bio-Rad公司。普通PCR仪购于美国Bio-Rad公司。生物安全柜购于美国Thermo Scientific公司。
1.3 方法
1.3.1 引物设计 在NCBI网站查找H37Rv标准菌株的全基因序列,并查找出Rv3480c基因,该基因全长1494bp,设计引物时,上游加入EcoR I酶切位点(下划线标记)、下游加入Hind III酶切位点(下划线标记),引物序列:Rv3480c-F-EcoRI(CCGGAATTCGTGAGCCAGACGGCCCGGC)和Rv3 480c-R-HindIII(CCCAAGCTTTCAGGAACCGAGGCCCGCG)。
1.3.2 目的基因扩增 以人型结核分枝杆菌标准菌株H37Rv DNA为模板,Rv3480c-F-EcoRI为上游引物,Rv3480c-R-HindIII为下游引物,使用Pfu高保真PCR反应试剂盒,在DNA扩增仪上进行目的基因的扩增。设置温度梯度摸索最佳反应条件,反应程序:预变性 94 ℃ 4 min,变性94 ℃ 30 s,退火58~70 ℃ 30 s,延伸72 ℃ 4 min,共34个循环,末次循环后补延伸72 ℃ 10 min,终止反应。琼脂糖凝胶电泳验证扩增条带,PCR产物经琼脂糖凝胶回收试剂盒回收纯化。
1.3.3 重组质粒的构建、扩增及表达 将目的基因和载体质粒分别进行EcoR I和HindIII双酶切,琼脂糖凝胶电泳后切胶回收酶切后的基因片段。酶切产物加入T4连接酶,置于PCR反应仪中,16 ℃,连接过夜。取10 μL连接产物转入感受态细胞DH5α中,涂布在含有卡那霉素的LB培养板上;37 ℃正置培养1 h,在倒置培养16~18 h;挑取单菌落接种于含有卡那霉素的LB培养液中培养过夜。质粒小抽提取DNA,双酶切验证。再送苏州金唯智生物科技有限公司完成测序验证。取10 μL验证构建成功的重组质粒转入BL21感受态细胞中,涂布于含有卡那霉素抗性的LB固体培养板上,37 ℃培养过夜。采用异丙基硫代半乳糖苷(IPTG),设诱导浓度梯度(1∶2 000;1∶5 000;1∶1 0000)16 ℃少量诱导表达,聚丙烯酰胺凝胶电泳(SDS-PAGE)验证,查找IPTG最佳诱导浓度。按最佳诱导浓度大量诱导表达后加入溶菌酶及苯甲基磺酰氟(PMSF);反复冻融后;在冰上超声(工作5s、间歇9 s、功率200 W)30 min,重复1次;加入100 μL DNase(100 μg/mL)(1∶500),4 ℃ 30 min;离心后验证诱导蛋白表达于上清中还是在包涵体中。
1.3.4 目的蛋白的纯化 经多次验证,诱导蛋白均显示表达于包涵体中。将超声破壁离心后的沉淀用IBS buffer(PH=8.0)洗3次,离心后弃上清;分别用6 M、4 M尿素(用1×PBS配制PH=8.0的6 M/4 M尿素)8 mL混匀;将8 mL分装至8个EP管内,-20 ℃冻存过夜;取出融化,10 000 g,2 min,将上清倒入裁剪好的透析膜中,两端封口;透析(2M尿素2L 6 h×2次、1 M尿素2L 6 h×2次、1×PBS 2L6h×2次)。保存纯化后的蛋白。SDS-PAGE,考马斯亮蓝染色初步验证。
1.3.5 Western Blot进一步验证 配制分离胶和浓缩胶、制备蛋白上样混合液,将配制好的分离胶和浓缩胶放入电泳槽中,加1×电泳液,上样孔分别加入蛋白上样混合液和蛋白质预染Marker,做好记录,80V电泳至Marker分层(约30 min),120 V电泳2 h;用半干转法将分离胶的目的蛋白转印至PVDF膜;用5 mL 5%脱脂奶粉封闭缓冲液封闭PVDF膜,再用0.1%PBST溶液洗膜3次,分别用一抗、二抗孵育,每次孵育后均用0.1%PBST溶液洗膜3次;最后用Bio-Rad凝胶成像仪曝光采集图像。
2 结果
2.1 Rv3480c基因扩增 以人结核分枝杆菌标准菌株H37Rv DNA为模板,Rv3480c-F-EcoRI为上游引物,Rv3480c-R-HindIII为下游引物扩增Rv3480c基因,设定退火温度梯度扩增后1%琼脂糖凝胶电泳验证,在6个温度下1494bp处均有较明显的条带,与目的基因大小相符,但61.4~68.2 ℃扩增均有杂带并随温度增高而减少(见图1A),以此得到最佳反应程序:预变性 94 ℃ 4 min,变性 94 ℃ 30 s,退火 70 ℃ 30 s,延伸72 ℃ 4 min,共34个循环,末次循环后补延伸72 ℃ 10 min,终止反应。
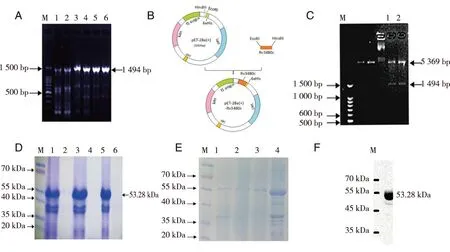
A:PCR扩增产物琼脂糖凝胶电泳结果M为500BP DAN marker,1-6分别是设定退火温度61.4 ℃、63.2 ℃、65.2 ℃、66.9 ℃、68.2 ℃、70.0 ℃;B:重组质粒pET-28a(+)-Rv3480c构建示意图; C:重组质粒pET28a-Rv3480c双酶切验证图 1、2为EcoRI和HindⅢ双酶切质粒pET28a-Rv3480c; D:pET28a-Rv3480c重组蛋白诱导表达后SDS-PAGE,M为蛋白分子量Marker,泳道1、3、5分别是1∶2 000、1∶5 000和1∶10 000的IPTG诱导沉淀,泳道2、4、6分别是1∶2 000、1∶5 000和1∶10 000的IPTG诱导上清; E:pET28a-Rv3480c重组蛋白SDS-PAGE,M为蛋白分子量Marker,泳道1为4M尿素溶解pET28a-Rv3480c重组蛋白,泳道2和3为4M尿素溶解pET28a-Rv3480c重组蛋白并纯化,泳道4为6M尿素溶解pET28a-Rv3480c重组蛋白; F:Rv3480c纯化后Western-blot鉴定胶图,M为标准物相对分子质量。图1 结核分枝杆菌包涵体蛋白Rv3480c的克隆和纯化
2.2 重组质粒 将扩增的目的基因和载体质粒pET28a分别进行EcoR I和HindIII双酶切后连接(见图1B),转化后的感受态细胞提取质粒并双酶切验证,可以看到在载体5369bp处和目的基因1494bp处均有条带(见图1C),重组质粒构建成功。送测序的重组质粒结果与人型结核分支杆菌标准菌株H37Rv基因序列比对显示重组质粒插入序列完全匹配。
2.3 抗原Rv3480c的诱导表达和纯化 将验证成功构建的重组质粒转入BL21感受态细胞中进行诱导表达,破菌体后进行SDS-PAGE跑胶验证,如图1D所示。结果显示1∶5 000和1∶10 000IPTG诱导浓度均可,且该蛋白表达于包涵体中。使用IBS buffer清洗,分别用6M、4M尿素溶解包涵体,并用低浓度尿素和PBS透析包涵体蛋白后,4M尿素溶解所得目的蛋白如泳道2和3所示,见较细单一目的条带。而6M尿素溶解所得目的蛋白较粗,且杂带较多图1E泳道4所示。
2.4 Western Blot验证表达的Rv3480c蛋白 经过SDS-PAGE,转膜、封闭、洗膜、抗His一抗孵育、再次洗膜、二抗孵育、显色等步骤,可见膜上出现特异性条带,如图1F。
3 讨论
结核病在全球蔓延的趋势不容忽视,控制结核病的关键之一在于早期诊断技术的革新。近年来许多新技术如流式细胞计数、质谱分析、蛋白芯片、血清学诊断等,都致力于寻找活动性结核病的生物标志物。发现新的活动性结核病生物标志物用于结核病诊断和病情监测已经成为当前研究重点[5]。
本研究目的蛋白Rv3480c是一种假定甘油二酯酰基转移酶,为结核分支杆菌分泌蛋白,是本课题组通过结核分枝杆菌蛋白芯片杂交技术筛选出的与活动结核病相关的蛋白。Daniel等[6]报道Rv3480c可能参与了结核菌休眠状态下的能量供应。结核分枝杆菌首次感染宿主后,激活宿主免疫反应,大部分宿主体内的细菌生长受到抑制而进入休眠状态,可在机体休眠数年甚至可达数十年。当宿主免疫低下时,细菌可复苏,宿主发展成为活动结核病。现有报道Rv3480c蛋白的英文文献仅两篇[8-9],中文未见报道,其分子结构以及生物学特性有待进一步的深入研究。
近几十年来生物基因工程技术得到了长足发展,基因克隆技术已被广泛应用于生物学各个领域,其中就包括结核病的研究。大肠杆菌为最常用的原核表达体系,该体系具有耗时短、经济、高效、操作简便等优点,但是该表达产物容易形成非水溶性、无生物活性的包涵体。本研究通过构建Rv3480c基因大肠杆菌表达体系表达出的蛋白几乎均存在于包涵体中,故需要溶解包涵体、蛋白变性和复性才能得到有生物活性的表达蛋白。本研究先经溶菌酶处理法破坏大肠杆菌细胞壁,再结合反复冻融法及超声破壁法充分裂解细菌,因超声波处理过程中产生大量热能,会使蛋白变性失活,为防止高温蛋白变性,操作需在冰上进行,间歇开机,而且操作时间不宜过长。为能够充分裂解细菌,我们超声破壁处理了2次,蛋白液出现黑色沉淀,分析可能是由于超声功率过大、操作时间过长导致蛋白炭化,在后续的实验中只超声处理1次,未再出现该情况。尿素作为包涵体溶解剂有很多优势,如成本低、容易透析清除、不电离、可直接进行SDS-PAGE检测等,所以我们选择尿素溶解包涵体,它可通过离子间的相互作用使包涵体蛋白质分子内、分子间的各类化学键断裂。我们首先应用6M尿素进行溶解包涵体,再使用透析复性法,通过逐步降低透液浓度来控制尿素清除速度,获得可溶性蛋白体积无明显变化,但是复性后蛋白杂质经SDS-PAGE显示有杂带,且蛋白液出现白色沉淀,考虑可能是透析复性过程中去除尿素速度过快。改用4M尿素后未出现沉淀,且经SDS-PAGE可见单一目的条带,获得可溶性蛋白液量不及6M尿素溶解所得。由此可见用低浓度尿素溶解包涵体得到的可溶性蛋白纯度高于高浓度尿素溶解,这与文献报道一致,因为变性剂浓度高时胞质颗粒蛋白也被释放出来[10],而且高浓度变性剂能够更充分地溶解包涵体,获得更多的可溶性蛋白。在透析复性过程中应缓慢去除变性剂,若速度较快,蛋白离子环境改变较大,容易形成沉淀,为后续纯化增加难度。
本研究采用原核表达体系成功克隆和纯化了结核分枝杆菌包涵体蛋白Rv3480c,为进一步进行结构和功能研究奠定了基础,也为更好地获取包涵体蛋白提供了新方法。
[1] 端木宏谨.我国结核病流行及其趋势[J].中国抗生素杂志,2004,29(12):732-734.
[2] WHO.Global Tuberculosis Report 2016[M].World Health Organization,2016.
[3] 王霄,文强,岳健博,等.GeneXpertMtb/RIF、抗酸染色及培养在浅表淋巴结结核诊断中的比较研究[J].遵义医学院学报,2016,39(3):275-278.
[4] Goletti D,Petruccioli E,Joosten S A,et al.Tuberculosis biomarkers: from diagnosis toprotection[J].Infectious Disease Reports,2016,8(2):24-32.
[5] 容豫,汤子健,曾珍,等.脑膜病变的MRI表现及诊断价值[J].遵义医学院学报,2015,38(5):512-515,522.
[6] Chou C H,Huang Y T,Hsu H L,et al.Rapid identification of the Mycobacteriumtuberculosis complex by an enzyme-linked immunosorbent assay[J].International Journal ofTuberculosis & Lung Disease,2009,13(8):996-1001.
[7] Kunnathvelayudhan S,Salamon H,Wang H Y,et al.Dynamic antibody responses to theMycobacterium tuberculosis proteome[J].Proceedings of the National Academy of Sciences of the United States of America,2010,107(33):14703-14708.
[8] Daniel J,Deb C,Dubey V S,et al.Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture[J].Journal of Bacteriology,2004,186(15):5017-5030.
[9] Deb C,Lee C M,Dubey V S,et al.A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded,drug-tolerant,dormant pathogen[J].Plos One,2009,4(6):e6077.
[10]Clark E D.Protein refolding for industrial processes[J].Current Opinion in Biotechnology,2001,12(2):202-207.
(编辑:王福军)
Cloning,expression and purification of the Rv3480c inclusion body protein of Mycobaterium tuberculosis
Wen Qiang1,2,Tang Mingmei1,Chen Ling1,Peng Zhangli1,He Yuejuan1,Tang Zhenghong1
(1.The Second Department of Respiratory Medicine,Affiliated Hospital of Zunyi Medical University,Zunyi Guizhou 563099,China; 2.The Third Department of Internal Medicine,Eastern Medical District of Linyi People’s Hospital,Linyi Shandong 276000,China)
ObjectiveTo clone,express and purify the Rv3480c inclusion body protein ofMycobateriumtuberculosisby molecular biological technique.MethodsRv3480c was amplified fromM.tuberculosisH37Rv by PCR and cloned into vector pET-28a,then transformed to E.coli BL21 (DE3) strain.The engineering bacteria were induced to express the protein by Isopropyl β-D-1-Thiogalactopyranoside (IPTG).The inclusion body was repeatedly dissolved,and recombinant protein was purified.ResultsThe gene that was amplified by PCR was successfully built in right sequence detected via agarose gel electrophoresis and sequencing.Recombinant protein was dialyzed and purified with urea solution and Phosphate Buffered Saline (PBS).The target protein was identified as Rv3480c by Western-blot,which was the recombination antigen ofM.tuberculosis.ConclusionThe purified Rv3480c protein was successfully obtained by molecular biological technique,which provides an experimental basis for further studies of the function and structure of Rv3480c,and explores a new method for better purification of inclusion body proteins.
Mycobacteriumtuberculosis; Rv3480c; inclusion body; cloning; purification
国家自然科学基金资助项目(NO:81360002)。
陈玲,女,博士,主任医师,硕士生导师,研究方向:肺部感染与结核病的防治,E-mail:lingjuncd@163.com。
R378.91
A
1000-2715(2017)04-0401-04
[收稿2017-06-04;修回2017-07-09]



