谭 煌,刘圣君,廖 刊
(1. 河北北方学院附属第一医院第一临床学院;2. 河北北方学院附属第一医院肾内科,河北 张家口 075000;3. 陆军军医大学第一附属医院,重庆 400000)
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是以朗格汉斯细胞克隆性增殖为特征的一组全身性罕见疾病,病因不明。LCH在任何年龄段都有可能发生,男女发病比例约为1.2∶1,小于15 岁儿童的年发病率约为4.6/100 万,成人年发病率约为1/100 万~2/100 万[1]。LCH 可累及全身各个脏器或系统[2],临床表现具有异质性,成人LCH一般诊断困难[3]。现报道我院1 例成人多系统LCH,旨在增加临床工作者对罕见疾病的系统认识,降低在临床工作中对该疾病漏诊、误诊、误治的可能性。
1 临床资料
患者,女性,55 岁,因颏下部肿物20 天于2016年6 月17 日入我院诊治。患者在过度劳累、精神紧张后逐渐出现颏下部肿物,伴疼痛、面部肿胀,就诊于当地医院,考虑“炎症”并给予克林霉素治疗5天,面部肿胀消退,但颏下部肿物仍存在,伴疼痛。患者为进一步诊治遂就诊于我院口腔科,查体可触及一枚大小约3 cm、质地中等、界限清楚的颏下部肿物,完善相关检查后行颏下部肿物切除术,术后病理(图1):(颏部肿物)考虑朗格汉斯细胞组织细胞增生症,结合临床病史符合嗜酸性肉芽肿;免疫组化(图2):S-100(+)、Ki-67(+50-75%)、CD68(+);患者术后伤口愈合佳,出院后定期返院复诊。2016 年10 月患者逐渐出现左侧耳前、右侧耳后及颏下淋巴结肿大(最大者约1.7 cm),四肢皮肤出现散在隆起硬结,局部色素沉着,伴瘙痒,患者自行口服中药(具体药物及用量不详)治疗后,皮疹消退,但仍间断出现,疗效欠佳。2016年11月患者为进一步治疗就诊于我院血液科,结合临床表现、病理检查结果和免疫组化诊断为LCH,行头颅X 线检查(图3):头颅骨骨缺损。于2016 年11 月至2017 年12 月使用COEP 方案(环磷酰胺1 g d1+长春新碱基2 mg d1+足叶乙甙100 mg d1-5+甲强龙80 mg d1-7)化疗共9个周期,同时给予唑来膦酸防止骨质破坏,化疗后患者肿大淋巴结较前明显缩小(最大者约1 cm)。2018 年4 月患者逐渐出现尿量增多,约每天12~16次,每次200~500 mL,每天总尿量约5 000 mL 左右,伴口渴、多饮。行头颅MRI 平扫(图4):矢状位垂体上缘膨隆,后叶高信号消失。垂体MRI 平扫(图5):垂体上缘轻度隆起,上下径最大约6.3 mm,垂体柄较居中。尿比重降低,肾功能未见异常,进一步完善禁水加压实验,考虑中枢性尿崩症,给予去氨加压素治疗后尿量较前明显减少,于2018 年4月给予COEP 方案化疗1 个周期。患者出院后多次就诊于北京协和医院门诊,查BRAF基因为阴性,建议使用以克拉屈滨为主的化疗方案治疗,患者因经济原因拒绝,于2018 年6 月至2018 年12 月给予COAP方案(环磷酰胺1 g d1+长春新碱基2 mg d1+阿糖胞苷0.1 g d1-7+醋酸泼尼松40 mg d1-7)化疗共4个周期,复查垂体MRI 平扫+强化(图6):神经垂体未显示,垂体柄局部略粗,脑内微量脱髓鞘变性灶。于2019 年2 月至2021 年11 月给予COAP 方案化疗共10 个周期,每个化疗周期均顺利结束。患者有高血压病病史多年,一直口服吲达帕胺控制,血压控制平稳。2021 年11 月入院查体无异常。辅助检查结果如上述。患者目前一般情况可,但尿量仍多,继续给予去氨加压素治疗,使用COAP 方案定期返院化疗中。
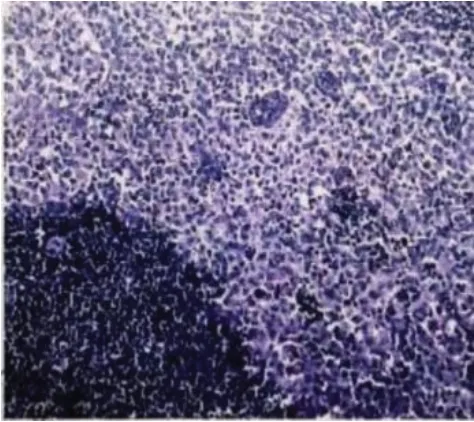
图1 术后病理(HE×100)
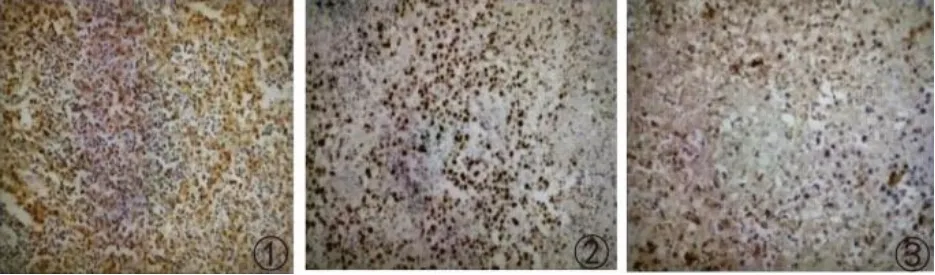
图2 免疫组化(×100)
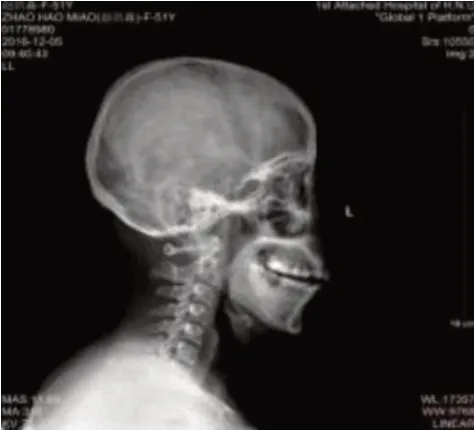
图3 头颅X线:颅骨骨缺损
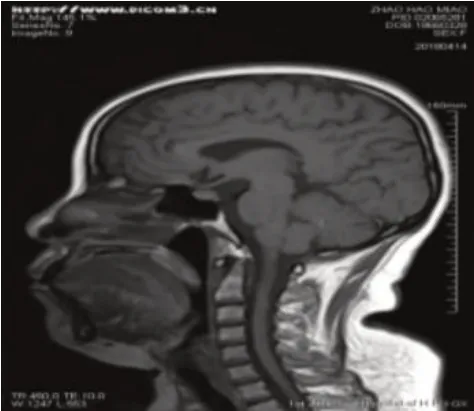
图4 头颅MRI平扫:垂体后叶高信号消失
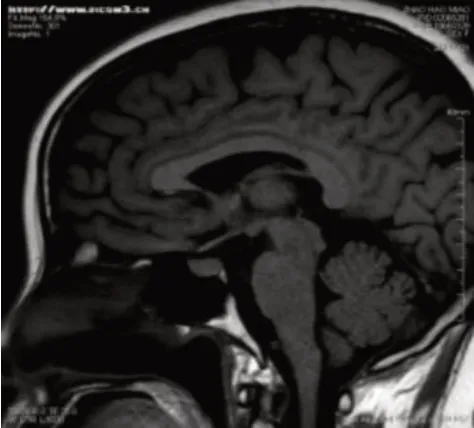
图5 垂体MRI平扫:垂体上缘轻度隆起
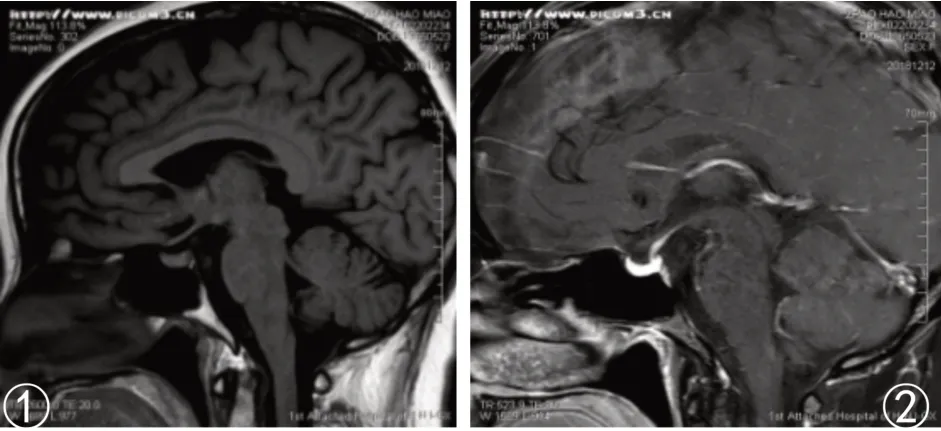
图6 垂体MRI平扫+增强
2 讨论
2.1 病因LCH 是一组以不成熟的树突状细胞克隆性增生性为特征的疾病,病因尚不明确,KHODDAMI M 等[4]报道,其病因可能与基因突变、病毒感染及环境等因素有关。关于LCH 是一种反应性还是肿瘤性疾病在学术界仍有争议[5],既往认为LCH是与免疫异常相关的疾病,近年来有研究[6]指出,LCH 是一种克隆性肿瘤性增殖,还有研究[7]指出,一半以上的LCH 患者存在BRAF V600E突变,而该基因突变可能是恶性肿瘤疾病的重要驱动因素,因此LCH 目前更倾向于是一种肿瘤性疾病。LCH 可以累及任何器官或系统,临床表现具有异质性,常见的累及部位有骨骼、肺、淋巴结、皮肤、中枢神经等脏器或系统[8]。
2.2 诊断病理检查结果联合免疫组化特异性的免疫标记物CD1α(+)、Langerin(CD207)(+)和S-100(+)是诊断LCH的金标准[6]。BRAF V600E基因突变可出现于LCH 的患者,诊断困难时,可以行基因检测辅助诊断[9]。有报道[2]指出,中枢性尿崩症可出现在LCH 确诊前,也可出现在确诊后,中枢性尿崩症的特征为垂体MRI 检查提示垂体后叶高信号消失、垂体柄增粗(>3 mm),再联合禁水加压素试验则能与其他原因导致的尿崩症相鉴别[10]。
2.3 治疗成人多系统LCH 目前无统一的标准治疗方案,具体治疗方案应根据病变部位及疾病严重程度个体化。多系统LCH 和高危器官(如骨髓、肝、脾)受累的单系统LCH 的治疗主要以化疗为主[11]。KOBAYASHI M 等[11]报道,对于多系统LCH,其一线治疗方案为长春新碱+甲泼尼龙,但目前一线治疗标准仍是经验性的方案,还有报道表明,对于累及多系统的难治性LCH,克拉屈滨+阿糖胞苷联合应用可作为二线治疗方案[12]。当中枢神经系统受累时,病变的部位常不易彻底修复,易遗留后遗症(如尿崩症等)影响到患者的生活质量,一般需要二线治疗[13],而大多数患者出现垂体功能减退后,经化疗通常也是不能逆转的,一般需要终身替代治疗[14]。成人多系统LCH 的标准治疗方案及疗效仍有待进一步临床研究和观察。



