马艺萍,阿不都热合曼·米吉提,袁玉娟,吐尔孙阿依·依斯米提拉,阿卜拉江·艾合麦提,穆叶赛·尼加提
(1.新疆医科大学研究生院,乌鲁木齐 830000;2.喀什地区第二人民医院心血管内科,新疆喀什 844099;3.新疆维吾尔自治区人民医院急救中心,乌鲁木齐 830000)
心血管疾病(cardiovascular disease,CVD)是一种与内皮细胞(endothelial cell,EC)水平相关的疾病[1]。EC损伤、血小板黏附和循环血液中的白细胞侵袭内皮下层是动脉粥样硬化形成的关键早期事件。内皮祖细胞(endothelial progenitor cell,EPC)是一种多功能干细胞,具有向EC分化的潜能[2]。当组织缺血或损伤时,EPC从骨髓被动员到外周血中进行分化。研究发现,EPC能有效减轻心绞痛和心力衰竭,改善左心室射血分数,并显示出组织器官再生和恢复缺血相关器官功能的前景[3]。微粒(microparticles,MPs)是由各种活化和凋亡细胞产生、直径为100~1 000 nm的小膜囊泡[4-6]。MPs是细胞间物质交换和信号转导的重要介质,携带表面抗原、蛋白质和各种生物活性分子,在细胞通讯中发挥关键作用[7]。有研究发现,EPC来源MPs可以保护心肌细胞免受血管紧张素-Ⅱ诱导的肥大和凋亡[8],并将微小RNA转移到EPC和EC,修复CVD中的组织损伤[9]。WU等[10]发现miR-126通过细胞外调节蛋白激酶(extracellular signal-regulated kinase,ERK)1/2信号通路来增强氧化应激下EPC的生物学功能,这可能为急性心肌梗死的治疗提供新的策略。然而,损伤的EPC来源MPs对EPC的作用及具体作用机制尚缺乏深入研究,因此,本研究通过体外实验,损伤处理EPC,提取MPs对EPC进行干预,为减少细胞组织损伤,开发阻断损伤EPC来源MPs试剂盒提供理论依据,现报道如下。
1 材料与方法
1.1 一般材料
EPC购于武汉普诺赛生命科技有限公司;异硫氰酸荧光素(fluorescein isothiocyanate,FITC)-CD34(11-0349-42)购于美国eBioscience公司;内皮细胞生长培养基(endothelial cell growth medium,EGM)-2(Promocell,C22121)、胎牛血清(Wisent,086-150)及青链霉素(Wisent,450-201-EL)均购于北京毕特博生物技术有限责任公司;肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)重组蛋白购于上海泽叶生物科技有限公司。检测设备:AcurriC6流式细胞仪购于美国BD公司;JEM1011透射电镜仪购于日本JEOL公司;Multiskan 51119000酶标仪购于美国Thermo fisher公司;Ts2R倒置显微镜购于日本Nikon公司);Veriti PCR仪购于美国Applied Biosystems公司;Mini Protean 3十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)仪器购于美国Bio-Rad公司;化学发光成像分析仪购于上海天能生命科学有限公司。
1.2 方法
1.2.1细胞培养
EPC为贴壁细胞,培养于EGM-2中,含有10%胎牛血清,100 U/mL的青霉素,100 μg/mL链霉素,放置于37 ℃、5% CO2的细胞培养箱中。当细胞长至80%~90%时,去除培养基,加入磷酸盐缓冲液(PBS)洗1~2次,去除PBS后,加入1 mL胰酶消化1~3 min后,加入3 mL完全培养基中和胰酶终止消化,将消化的细胞转移至15 mL离心管中,1 000 r/min离心5 min。倒掉上清液,加入3 mL培养基重悬细胞,1∶3传代至培养皿中培养。
1.2.2流式细胞仪检测
取分离培养的EPC,去除培养基,加入PBS洗1~2次,去除PBS后加入1 mL胰酶消化1~3 min,加入3 mL完全培养基中和胰酶终止消化,将消化的细胞转移至15 mL离心管中,1 000 r/min离心5 min。倒掉上清液后,加入3 mL PBS重悬细胞。加入PBS-1%牛血清白蛋白(BSA)重悬细胞,加入CD34抗体按照1∶50的比例稀释,空白对照细胞加入同型对照抗体,冰上孵育30 min。细胞中加入400 μL PBS混匀,1 000 r/min离心,去除上清液,加入PBS洗3次,加入500 μL PBS缓冲液重悬细胞。CD34是EPC的特征性分子标志物,采用流式细胞仪检测分离细胞中的CD34阳性细胞群,见图1。
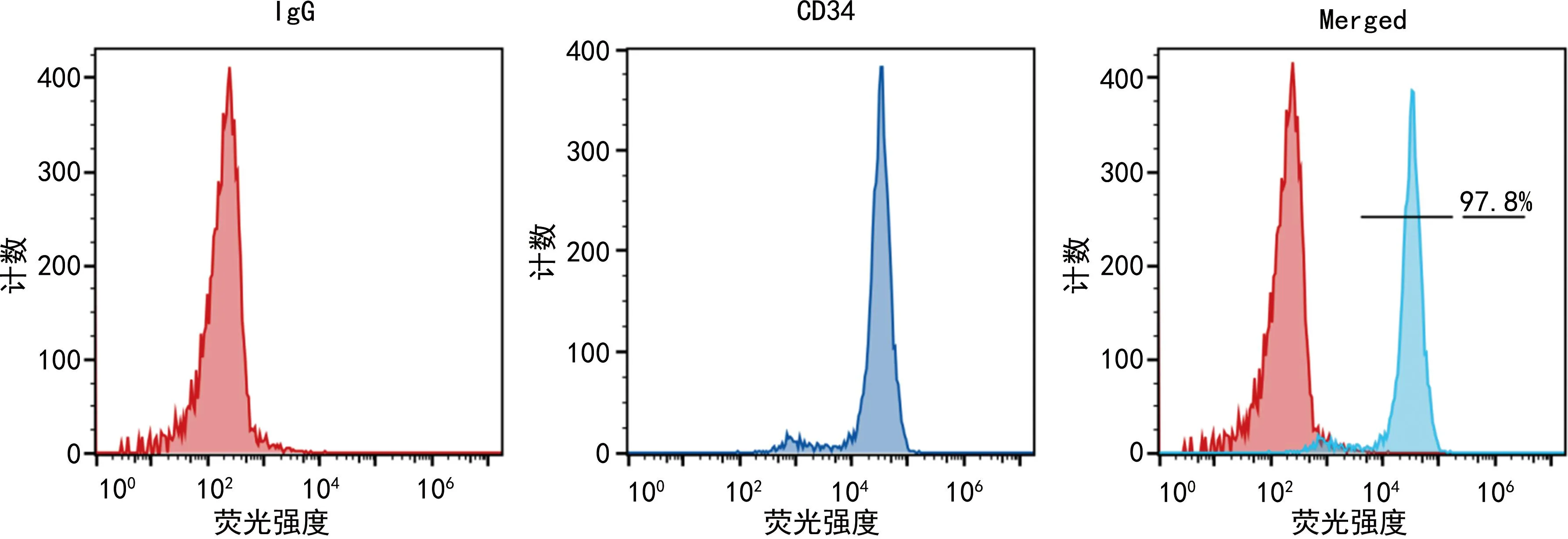
图1 EPC流式鉴定
1.2.3HG、TNF-α损伤处理
将EPC培养于培养皿中,待细胞贴壁后分别进行正常培养、高糖(high glucose,HG)培养、加入TNF-α重组蛋白培养24 h后收集上清液并提取MPs,此为HG、TNF-α损伤的EPC来源MPs。将1×106个正常EPC接种于6孔板中,待细胞贴壁后,将50 μg/mL正常培养EPC来源MPs(control-EPC-MPs组)、50 μg/mL HG培养EPC来源MPs(HG-EPC-MPs组)、50 μg/mL TNF-α培养EPC来源MPs(TNF-α-EPC-MPs组)分别加入正常培养的EPC后,收集细胞进行检测。
1.2.4透射电镜检测
3组细胞用胰酶消化后,收集到离心管中,2 000 r/min离心5 min,去除培养基。向收集的细胞中加入戊二醛稀释液固定备用;取5~10 μL细胞悬液加到Formvar-carbon载样铜网上;取100 μL PBS加到封口膜上,用镊子将铜网(Formvar膜面朝下)放在PBS液滴上清洗。在所有步骤中,保持Formvar膜面湿润而另一面干燥;铜网放在50 μL 1%戊二醛液滴上5 min;铜网放在100 μL ddH2O洗2 min(洗8次);铜网放在50 μL草酸双氧铀液滴上(pH 7.0)5 min;铜网放在50 μL甲基纤维素液滴上10 min,冰上操作;铜网放到样品台顶端的不锈钢环上,在滤纸上吸去多余液体。空气干燥5~10 min;铜网放在样品盒里,拍摄透射电镜照片。
1.2.5MTT检测
3组细胞培养24 h后,加入10 μL MTT(5 mg/mL),在细胞培养箱中继续培养3 h。去除培养基,每孔加入100 μL Formazan溶解液,适当混匀,在细胞培养箱内继续孵育,直至普通光学显微镜下观察发现Formazan全部溶解。37 ℃孵育4 h,紫色结晶全部溶解。如果紫色结晶较小、较少,溶解时间较短;如果紫色结晶较大、较多,溶解时间延长,可适当振摇数次。在570 nm测定吸光度(A)值;如无570 nm滤光片,可使用560~600 nm滤光片。
1.2.6细胞划痕实验
将5×106个EPC接种于6孔板中,摇晃均匀后放入培养箱中培养。待细胞长满后,去除培养基,用PBS洗1次;200 μL枪头在6孔板中划3道直线,PBS洗2次,去掉划出的细胞;用PBS洗2次,将残余的细胞洗干净;在显微镜下拍照,加入无血清培养基,再分别加入control-EPC-MPs组、HG-EPC-MPs组、TNF-α-EPC-MPs组细胞,摇晃均匀后放入培养箱中常规培养12、24 h后拍照。
1.2.7细胞管腔形成实验
3组细胞用胰酶消化,1 000 r/min离心,去除上清液,加入完全培养基重悬细胞,将细胞分别种在24孔板中,每孔种1×105个细胞。培养6 h后,观察EC管腔形成情况并拍照。拍照后,使用Image J软件Angiogenesis Analyzer Plugin插件对管腔形成情况进行分析。
1.2.8实时荧光定量PCR(qPCR)检测
3组细胞去除培养基,加入PBS洗1次,加入1 mL TRIzol裂解细胞,将裂解液转移至EP管中。加入氯仿200 μL,用力振摇15 s,室温静置15 min,4 ℃ 12 000 r/min离心15 min。离心后液体分为3层,小心吸取上层无色液体移入1个新的EP管中。加入等体积异丙醇,上下颠倒混匀,室温静置5 min,4 ℃ 12 000 r/min离心10 min。去除上清液,沉淀加入75%乙醇1 mL,轻微振荡15 s,4 ℃ 7 500 r/min离心5 min。小心去除上清液,管内沉淀在超净台中鼓风静置干燥3~5 min。加入50 μL DEPC水溶解,nanodrop检测浓度水平,-80 ℃冰箱保存。RNA纯度根据nanodrop检测结果和230、260、280 nmA值决定。内皮型一氧化氮合成酶(endothelial nitric oxide synthase,eNOS)、沉默信息调节因子1(silent information regulator 1,SIRT1)、ERK、大鼠肉瘤(rat sarcoma,RAS)引物情况见表1。

表1 PCR检测引物序列
1.2.9Western blot
3组细胞加入200 μL RIPA裂解液,冰上裂解30 min。裂解后将裂解产物转移到预冷的1.5 mL离心管(冰上)并在4 ℃下以12 000 r/min离心10 min。将离心后的上清液转移至1.5 mL离心管中,-20 ℃保存。采用BCA法检测总蛋白水平,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate,polyacrylamide gel electrophoresis,SDS-PAGE)分离蛋白。浓缩胶黏剂层电压为80 V,电泳时间为30 min。样品进入分离胶黏剂后,在120 V下电泳90 min,然后将蛋白转移到硝酸纤维素膜上。在脱脂乳中封闭后,将膜与一抗在4 ℃孵育过夜。根据一抗,将膜与二抗在25 ℃孵育2 h。使用化学发光成像分析仪进行扫描成像。
1.3 统计学处理

2 结 果
2.1 3组EPC电镜下细胞器变化
control-EPC-MPs组细胞微管结构完整,整体较长,高尔基体结构完整;HG-EPC-MPs组细胞微管结构完整,长度无明显差异,高尔基体结构相对完整;TNF-α-EPC-MPs微管结构完整,长度有所缩短,高尔基体结构相对完整,部分高尔基体出现囊泡状散在分布在细胞质中,见图2。

绿色箭头:微管;蓝色箭头:高尔基体。
2.2 3组EPC细胞增殖情况比较
与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组细胞增殖率明显下调,差异有统计学意义(P<0.01),见图3。

①:control-EPC-MPs组;②:HG-EPC-MPs组;③:TNF-α-EPC-MPs组;a:P<0.01,与control-EPC-MPs组比较。
2.3 3组EPC细胞迁移情况比较
与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组0 h细胞迁移能力无明显变化(P>0.05),12、24 h细胞迁移能力明显降低,差异有统计学意义(P<0.01),见图4。
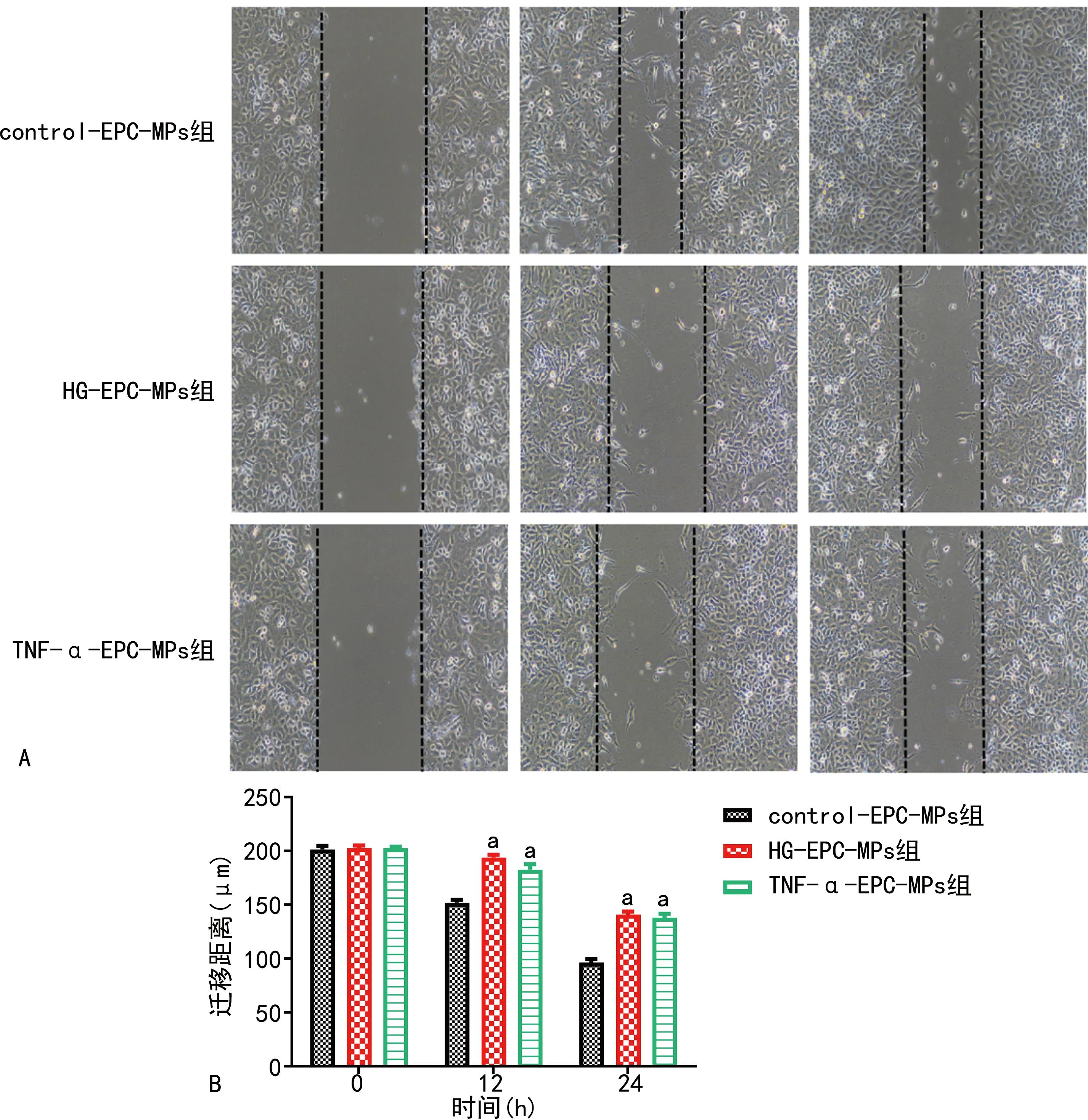
A:细胞划痕图(100×);B:3组EPC迁移距离比较;a:P<0.01,与control-EPC-MPs组比较。
2.4 3组EPC管腔形成情况比较
与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组成管明显减少,差异有统计学意义(P<0.01),见图5。

A:管腔形成图(100×);B:成管分析结果;①:control-EPC-MPs组;②:HG-EPC-MPs组;③:TNF-α-EPC-MPs组;a:P<0.01,与control-EPC-MPs组比较。
2.5 3组EPC的mRNA和蛋白表达水平比较
与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组eNOS、SIRT1、RAS和ERK的mRNA和蛋白表达水平下调,差异有统计学意义(P<0.05),见图6~7。

A:3组eNOS mRNA表达比较;B:3组SIRT1 mRNA表达比较;C:3组RAS mRNA表达比较;D:3组ERK mRNA表达比较;①:control-EPC-MPs组;②:HG-EPC-MPs组;③:TNF-α-EPC-MPs组;a:P<0.05,与control-EPC-MPs组比较。

A:3组eNOS、RAS Western blot图;B:3组SIRT1、ERK Western blot图;C:3组eNOS蛋白表达分析;D:3组SIRT1蛋白表达分析;E:3组RAS蛋白表达分析;F:3组ERK蛋白表达分析;①:control-EPC-MPs组;②:HG-EPC-MPs组;③:TNF-α-EPC-MPs组;a:P<0.05,与control-EPC-MPs组比较。
3 讨 论
从骨髓动员进入循环的EPC对血管修复、管壁组织重塑和血管生成[11]具有重要意义。前期研究发现,MPs在CVD的发生、发展中发挥极其重要的作用[12]。研究人员已经证实,EPC来源MPs可以修复CVD中的组织损伤[9-10]。本研究应用HG和TNF-α介导EPC损伤来源MPs,探索EPC功能障碍的机制,这可能为将来阻断EPC损伤来源MPs、减少EPC损伤提供理论依据。
本研究发现,HG和TNF-α介导EPC损伤来源MPs下调了EPC细胞增殖,降低了细胞迁移能力,细胞成管明显减少;HG-EPC-MPs组和TNF-α-EPC-MPs组EPC损伤加重,并引起EPC微管和高尔基体等细胞器轻微改变;HG-EPC-MPs组和TNF-α-EPC-MPs组EPC变化可能与SIRT1/ERK1信号通路有关。有研究强调了线粒体和内质网在维持内皮功能中的核心作用[13]。然而,关于微管和高尔基体在内皮功能中的作用鲜有报道。本研究发现,HG-EPC-MPs组、TNF-α-EPC-MPs组的EPC微管结构完整,长度有所缩短,高尔基体结构相对完整,部分高尔基体出现囊泡状散在分布在细胞质中,提示HG和TNF-α介导EPC损伤来源的MPs可能会导致微管和高尔基体结构改变,从而加重应激损伤反应。因此,高尔基体损伤可能在EPC的损伤中发挥重要作用。
SIRT1是哺乳动物NAD依赖的sirtuin脱乙酰基酶组成部分,影响炎症、线粒体生物发生、细胞衰老和凋亡等多个过程[14-15]。CAO等[16]证明了原儿茶酚醛通过调节SIRT1诱导自噬和抑制凋亡来挽救氧糖剥夺/再灌注(oxygen-glucose deprivation/reperfusion,OGD/R)诱导的EC损伤。此外,LI等[17]提出miR-126过表达通过促进SIRT1/Nrf2信号通路来减轻氧化应激和炎症反应,从而减轻OGD/R诱导的人脐静脉EC(human umbilical vein endothelial cells,HUVECs)的神经毒性。本研究中,与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组SIRT1的mRNA和蛋白表达水平下调。此外, HG-EPC-MPs组和TNF-α-EPC-MPs组的EPC增殖率、细胞迁移能力降低,细胞成管明显减少。BI等[18]证明,血管生成素-1通过Tie2受体/ERK1/2-p38MAPK通路减轻了肾小球EC中内质网应激诱导的细胞功能障碍和凋亡。WANG等[19]研究表明,碱性成纤维细胞生长因子参与了缺血氧化损伤模型中内质网应激和线粒体功能障碍的抑制,这是PI3K/Akt和ERK1/2信号通路激活的基础。与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组ERK1的mRNA及其蛋白表达下调,表明HG和TNF-α介导EPC损伤来源的MPs可以通过影响SIRT1/ERK1的表达和改变高尔基体结构来抑制EPC的生物学功能。
eNOS是一种产生NO的酶,在心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)中具有重要意义。CAPE-oNO2通过SIRT1/eNOS通路抑制氧化应激、炎症反应和细胞坏死,从而减轻MIRI[20]。PENG等[21]证明CC基元配体2促进MAPK/ERK1/2/MMP-9、PI3K/AKT和Wnt/β-catenin信号通路相关蛋白的表达,并以浓度依赖方式促进HUVECs的增殖、迁移和管形成。醛糖还原酶通过SIRT1/AMPK-α1/mTOR通路抑制HG诱导的eNOS和血管细胞黏附因子1表达,调控单核细胞白血病单核细胞与HUVECs的黏附,从而调控HG诱导的HUVECs死亡[22]。本研究中,与control-EPC-MPs组比较,HG-EPC-MPs组、TNF-α-EPC-MPs组SIRT1的mRNA和蛋白表达水平下调。RAS的激活在MIRI的发生、发展中发挥重要作用[23]。当MIRI发生时,肾素-血管紧张素-醛固酮系统的最终代谢产物血管紧张素Ⅱ与血管紧张素Ⅰ型受体结合,刺激线粒体氧化应激损伤反应,导致活性氧过度产生[24]。活性氧过度产生可能诱发强烈的炎症反应,增加心肌梗死面积,导致心功能异常。LI等[25]研究表明,高尔基体参与了钙超载的形成和缓解,并对神经细胞的缺血和再灌注有强烈的反应。因此,本研究证实HG和TNF-α介导EPC损伤来源MPs可能通过影响EPC中eNOS、RAS的表达,促进氧化应激、炎症反应和细胞坏死,从而加重EPCs的损伤。
综上所述,HG和TNF-α介导EPC损伤来源MPs可能通过调控SIRT1/ERK1通路蛋白的表达,使EPC内含细胞器的结构改变,从而影响EPC的生物学功能。



