孙键,毛星星,龙圆圆,沙梦琪,刘喜平,陈艳*,黄佩*
葡萄糖-6-磷酸脱氢酶(G6PD)作为磷酸戊糖途径(PPP)的关键酶,通过调节核糖-5-磷酸(R-5-P)和还原型辅酶Ⅱ(NADPH)的生成,在核苷酸和脂质的生物合成以及维持氧化还原的动态平衡中起着至关重要的作用。G6PD的活性在快速增殖的细胞中明显升高,适当增强G6PD表达可以延长转基因小鼠的寿命,严重的G6PD缺乏常导致胚胎发育缺陷甚至死亡[1-4]。现有研究表明,G6PD表达及活性异常不但与自噬、胰岛素抵抗、感染、炎症以及糖尿病、高血压等多种病理生理及疾病的发生、发展有关[5],也与肿瘤的发生、发展密切相关。本文总结了肿瘤中G6PD参与的代谢反应及与G6PD表达、活性相关的信号通路的研究成果,并对G6PD是否能作为潜在的抗肿瘤治疗靶点进行 了讨论。
1 G6PD与肿瘤密切相关
肿瘤的发生是一个动态而复杂的过程,能量及代谢的改变是肿瘤的基本特征之一[6]。正常细胞中,葡萄糖通过糖酵解生成丙酮酸,丙酮酸转化为乙酰辅酶A后,进入线粒体参与三羧酸循环(TCA)[7],最终通过氧化磷酸化生成三磷酸腺苷(ATP),同时也为脂质和非必需氨基酸的合成提供了中间代谢产物[8]。然而,肿瘤细胞对上述代谢途径进行了重新编程[9]。代谢的重新编程导致肿瘤细胞获得能量的主要方式变为有氧糖酵解(Warburg效应)[10]。葡萄糖在进入细胞后被己糖激酶磷酸化为葡萄糖-6-磷酸(G-6-P),并经过一系列反应生成丙酮酸,最终可通过糖酵解获得能量。大量的糖酵解代谢中间产物也促进了PPP,PPP包括氧化分支和非氧化分支。在PPP的氧化分支中,G-6-P被氧化代谢生成R-5-P和NADPH;在非氧化分支中,PPP则是以糖酵解代谢中间产物为原料进行可逆反应,其主要作用是生成R-5-P[11]。
G6PD作为PPP氧化分支的限速酶,参与调节R-5-P和NADPH的合成[11]。R-5-P不仅是核苷酸合成的重要成分,也是细胞内多种生物分子的合成前体,其中包括ATP、ADP、AMP、c-AMP、辅酶A、FAD、NAD(P)和NAD(P)H等。快速增殖的肿瘤细胞需要大量的R-5-P以维持其生长和生存所需的核苷酸及其他生物分子合成的需求。PPP的另一代谢产物,NADPH可以促进四氢叶酸(胸苷酸合成酶辅助因子)的生成及参与核苷酸还原酶的合成[12-13]。此外,脂质的合成同样也需要NADPH的参与,如胆固醇的合成等[14]。因此,G6PD调控R-5-P及NADPH的生成对于肿瘤细胞大分子物质的生物合成至关重要。G6PD主要参与的肿瘤生物学过程见图1。
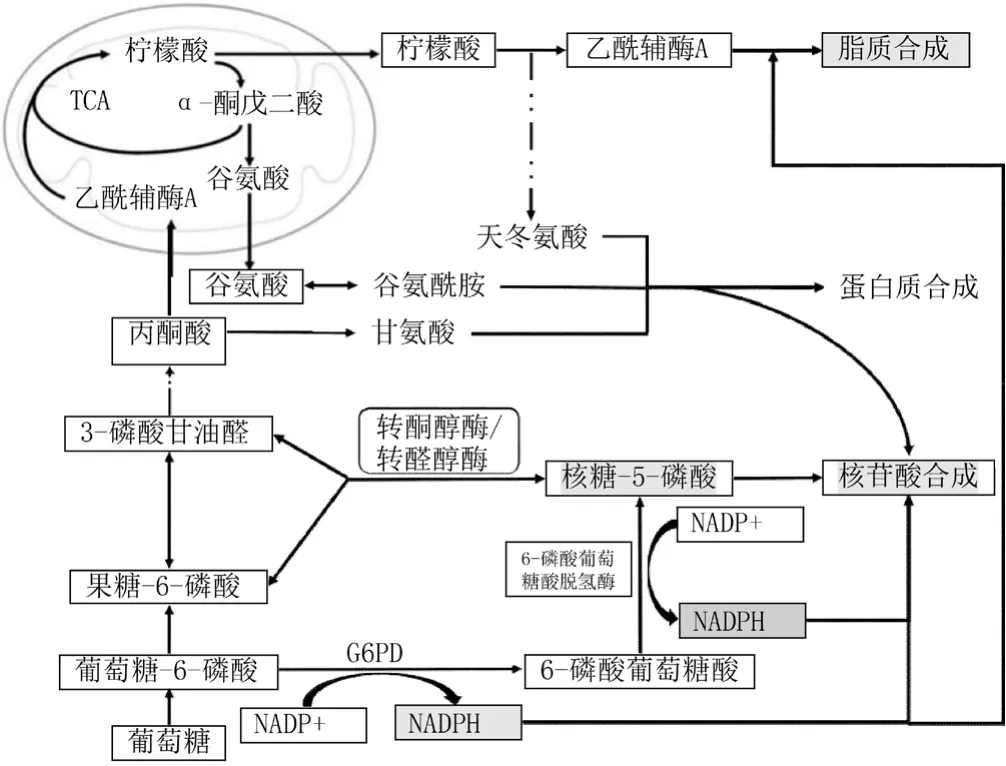
图1 G6PD主要参与的肿瘤生物学过程Figure 1 Biological processes that G6PD mainly involved in tumor
NADPH还能通过维持谷胱甘肽(GSH)的还原形态,影响GSH对活性氧(ROS)的解毒功能,而ROS解毒功能在抗氧化应激的反应中起着核心作用。ROS对细胞具有多重作用[15],如低水平ROS可通过激酶与磷酸水解酶的转录后修饰使细胞存活,进而促进正常细胞的增殖[16]。肿瘤细胞由于异常的能量代谢以及过量的生物合成等,造成ROS水平升高,诱导DNA损伤,促进促癌基因的激活[17-19];若ROS水平过高,超过肿瘤细胞的负荷阈值,又会导致细胞死亡[20-22]。肿瘤细胞可以通过G6PD对NADPH的合成进行调节,保护肿瘤细胞及生物大分子物质免受ROS的损害,使其在高ROS水平下存活。在更注重维持自身氧化还原反应稳定的细胞中,R-5-P通过糖异生合成G-6-P,G-6-P经PPP的氧化分支生成NADPH;而在快速增殖的细胞中,G-6-P则在糖酵解后通过PPP的非氧化分支生成R-5-P。G-6-P是否参与这两种反应主要取决于细胞对能量、生物合成及氧化还原平衡的需求。
因此,作为PPP限速酶的G6PD与肿瘤密切相关。肿瘤细胞中G6PD的调控,不仅依赖于其转录及活性,也与翻译后的进一步修饰有关。
本文价值:
(1)总结了G6PD在肿瘤生物合成及氧化还原中的作用,提升了临床对G6PD在肿瘤中作用的全面认识;(2)通过对肿瘤中G6PD的表达、活性及其相关调控通路、作用机制进行总结、归纳、分析,利于进一步研究其在不同类型肿瘤中的作用机制;(3)汇总了G6PD在多种肿瘤中的作用及机制,为靶向G6PD治疗肿瘤的相关研究提供了参考。
本文局限性:
(1)文献检索方法只选择了PubMed数据库,可能会遗漏部分相关研究;(2)由于人工筛选文献,可能会出现偏差;(3)未对G6PD抑制剂及其临床应用进行更深入的挖掘。
2 肿瘤中G6PD的调控及作用
2.1 肿瘤中G6PD表达的调控及作用 信号转导和转录激活因子(STAT)与G6PD的表达水平有关。STAT蛋白是参与免疫、细胞增殖、凋亡和分化的转录因子,其功能失调与肿瘤的发生有关[23]。在黑色素瘤中,STAT3和STAT5被高度激活,G6PD的敲除及过表达均会导致p-STAT3/5发生改变[24]。然而,G6PD与STAT3/5之间的调控机制尚不明确。相比于G6PD缺陷型裸鼠,野生型G6PD裸鼠的肿瘤生长速度更快,体积更大,且恶性程度更高[25],说明G6PD可能会影响黑色素瘤的增殖及分化。ZHANG等[26]发现,在肾癌中G6PD的表达会受到p-STAT3激活剂或抑制剂的影响。G6PD通过促进ROS的生成,导致p-STAT3和细胞周期蛋白D1(CyclinD1)表达增加。p-STAT3也可以通过与G6PD启动子结合从而激活G6PD的转录,这表明p-STAT3与G6PD的过表达形成了一个正反馈调控环。此外,在黑色素瘤中也发现G6PD与细胞周期蛋白(cyclin D1和cyclin E)的表达相关,并且在G6PD缺陷的黑色素瘤裸鼠模型中,抗凋亡蛋白Bcl-2和Bcl-xl的表达明显降低[25]。以上发现表明G6PD在STAT3/5介导的肿瘤细胞增殖、分化及凋亡中发挥着重要作用。
TAp73与G6PD的表达水平相关。TAp73作为P53的相关蛋白,不仅是糖酵解的关键调控因子[27],也参与了氧化应激的调节。TAp73通过促进肿瘤细胞的PPP,增加了核苷酸和氨基酸的合成[28],其机制可能与G6PD促进R-5-P及NADPH的合成有关。在肺癌中,TAp73通过与G6PD内含子特异性结合,激活G6PD的表达,促进肿瘤细胞增殖,而敲低TAp73会使G6PD mRNA水平明显降低,核苷酸合成减少。TAp73敲除的肿瘤细胞可以通过过表达G6PD或ROS清除剂来减轻敲低TAp73对自身增殖造成的影响[29-30]。这些研究表明了TAp73是维持肿瘤细胞内G6PD表达所必需的蛋白,其通过促进核苷酸的合成、维持氧化还原平衡来促进肿瘤细胞的增殖。WANG等[31]对膀胱癌的研究发现,应用RAS抑制剂或敲除TAp73也能得到上述类似的结果,说明阻断RAS信号通路可能会抑制TAp73,从而降低G6PD的表达。但在K-RAS驱动的肿瘤中,G6PD对肿瘤的生长和转移并不会起到关键作用[32]。
癌基因c-Myc参与G6PD表达的调控并在调节肿瘤细胞周期进程和能量代谢中起重要作用[33]。c-Myc参与了DNA结合抑制因子(ID1)诱导的G6PD激活。在ID1敲除的肿瘤细胞中,G6PD mRNA水平明显降低,NADPH降低,ROS水平升高;将G6PD或c-Myc转染ID1敲除的肝癌细胞会逆转上述表现。进一步研究表明,ID1通过Wnt /β-连环蛋白(Wnt/β-catenin)途径激活c-Myc,增强G6PD启动子的激活[34]。此外,长链非编码RNA-蛋白质二硫键异构酶家族A成员3假基因1(lncRNA-PDIA3P)也可以通过c-Myc增强G6PD转录,并通过PPP升高NADPH水平,从而促进多发性骨髓瘤的进展[35]。
磷脂酰肌醇3-激酶/蛋白激酶B(PI3K/AKT)在调控G6PD的表达上发挥重要作用[36]。SUN等[37]发现激活AKT通路可以上调G6PD表达、促进NADPH的合成、提高肿瘤细胞在氧化应激下的存活。CHENG等[38]发现PI3K/AKT通过抑制E3连接酶减少G6PD的降解,促进了代谢的重编程。磷酸酶和张力蛋白同源基因(PTEN)作为PI3K/AKT的负调控因子,通过糖原合酶激酶3β(GSK-3β)介导的磷酸化使T淋巴细胞白血病/淋巴瘤蛋白(TCL1)失活,抑制G6PD premRNA剪接,从而抑制PPP并最终减少能量生成及生物合成[39],并且抗PPP治疗也显著抑制了PTEN缺失的肿瘤和小鼠模型肿瘤的生长[38]。
抗氧化调节因子核因子E2相关因子2(NRF2)是肿瘤进展、转移和耐药的重要驱动因素[40]。ZHANG等[41]发现抑制NRF2可以降低G6PD的表达水平,其通过G6PD/缺氧诱导因子-1α(HIF-1α)上调Notch1的表达,从而影响Hes家族BHLH转录因子1(HES1)和上皮细胞-间充质转化(EMT),增强了乳腺癌细胞的增殖及迁移能力。由NRF2介导的G6PD表达上调也与胆管癌的顺铂耐药相关[42]。人端粒酶逆转录酶(TERT) 是端粒酶的组成成分之一[43],端粒酶在肿瘤中通常有较高的活性[44]。在神经胶质瘤中,NRF2可以促进TERT的表达及代谢的重新编程;抑制TERT的活性能通过ROS依赖的方式诱导细胞凋亡;经RNA干扰技术抑制TERT则降低了G6PD的表达[43]。上述研究表明,G6PD可能也参与了NRF2-TERT的抗氧化防御反应作用。
AMP依赖的蛋白激酶(AMPK)作为能量代谢调节的关键分子[45],可以通过G6PD调控肿瘤细胞的代谢重编程。肿瘤细胞可以通过激活AMPK增加NADPH的合成来对抗氧化应激[15]。YANG等[46]发现在乳腺癌中,高水平的ROS显著抑制了非锚定依赖性生长。G6PD作为ROS的主要调控因子,可以通过AMPK显著下调G6PD mRNA及蛋白水平。4-羟基苯丙酮酸双加氧酶(HPD)也可以经肝激酶B1- AMP依赖的蛋白激酶(LKB1-AMPK)信号介导的组蛋白脱乙酰基酶10(HDAC10)的磷酸化,增强G6PD表达,通过代谢重编程,降低ROS水平,进一步促进肺癌发展[47]。
非编码RNA也可以调控G6PD的表达。miR-1、miR-122、miR-206、miR-613可以通过靶向3'非编码区(3'UTR)下调G6PD mRNA水平,抑制鼻咽癌、食管癌、肝癌、宫颈癌等增殖、侵袭及转移[48-52]。WANG等[53]和 ZHAO等[54]发现lncRNA-OR3A4、SNHG14分别可以抑制miR-1207-5p、miR-206,从而促进G6PD表达,并通过促进R-5-P及NADPH的合成影响骨肉瘤、肺癌的发展。piRNA823也可以对G6PD/HIF-1α轴进行调节促进大肠癌细胞的增殖、侵袭[55]。肿瘤中 G6PD 表达的调控及作用见表1[25-26,30,32,34-35,38-39,41,43,46-67]。

表1 肿瘤中G6PD表达的调控及作用Table 1 Regulation and function of G6PD expression in tumors
2.2 肿瘤中G6PD活性的调控及作用 G6PD的活性与其在体内的存在形式有关,其既可以无催化活性或催化活性很低的单体形式存在,也可以催化活性高的二聚体形式存在,有些细胞甚至存在四聚体(包含两个二聚体)[68]。P53是肿瘤中最常见的突变基因之一[69],与TAp73不同的是,P53或其他P53家族成员的表达改变均不影响G6PD的转录[30]。实验证实,P53可以通过与G6PD结合,抑制G6PD二聚体的形成,从而影响葡萄糖的消耗及生物合成,降低NADPH水平[70]。那么,在发生P53突变的肿瘤中,G6PD的活性是否也发生了改变?P53为P21激活酶4(PAK4)的下游蛋白[71],在结肠癌中,PAK4过表达促进了葡萄糖和NADPH的生成,并且G6PD的活性升高与PAK4过表达有关,PAK4敲除降低了G6PD的活性[72]。这些研究表明,PAK4诱导葡萄糖消耗和生成NADPH是通过增强G6PD活性介导的。P53可以与PAK4相互作用,沉默PAK4能够增强P53蛋白的表达。MDM2也可以通过抑制P53的转录活性及促进P53的泛素化降解调控P53[73]。PAK4可以与MDM2相互作用,敲除 PAK4可以降低MDM2水平。PAK4可以促进MDM2与P53结合[72]。这些结果表明,PAK4能增强MDM2与P53的相互作用,通过促进P53降解减少P53与G6PD结合,促进G6PD二聚体的形成并增强G6PD的活性。此外,PTEN、Bcl-2相关永生基因3(BAG3)及生长抑制特异性基因5(GAS5)也能通过与G6PD蛋白结合,从而抑制G6PD的二聚体形成,降低G6PD活性,抑制肿瘤的发生、发展[39,74-75]。G6PD活性的调控及作用总结见表2[39,70,72,74-80]。

表2 肿瘤中G6PD活性的调控及作用Table 2 Regulation and function of G6PD activity in tumors
G6PD的活性还与其化学修饰有关。MA等[76]发现,有丝分裂的关键调节因子Polo样激酶(Plk1)能够促进G6PD的磷酸化,增加G6PD的活性,从而调控肿瘤的生长和细胞周期进程。此外,乙酰化修饰也参与了G6PD活性的调节。在白血病中,去乙酰化酶(SIRT2)通过对G6PD乙酰化的调节,参与AML的代谢重编程[77]。热休克蛋白27(HSPB1)也可以激活G6PD以对抗氧化应激或DNA损伤;HSPB1增强了G6PD与SIRT2的结合能力,导致G6PD去乙酰化及活化[78]。阿司匹林还可以通过G6PD乙酰化,降低G6PD活性,减少R-5-P和NADPH的合成,从而发挥抗结肠癌作用[79]。另外,抑制G6PD的糖基化会降低肺癌细胞在体内外的增殖、生长[80],其机制可能与核酸、脂质的生物合成以及抗氧化还原有关。
2.3 G6PD对肿瘤细胞死亡的作用 G6PD缺乏或活性降低易引起肿瘤细胞凋亡[5]。G6PD敲低的食管癌异位移植瘤生长速度减慢,且凋亡相关蛋白水平明显上升[61]。在具有凋亡抵抗特性的急性髓细胞白血病HL-60细胞中,G6PD水平相较于凋亡敏感的细胞更高[81]。G6PD引起的死亡也与自噬及铁死亡密切相关。自噬受氧化还原稳态和葡萄糖代谢的调节[82]。在乳腺癌相关研究中发现,通过抑制G6PD诱导的内质网应激,能导致不依赖mTOR信号通路的自噬小体形成,解除对肿瘤细胞自噬的调控,同时增强了拉帕替尼的细胞毒作用[59]。铁死亡是最新发现的一种调节细胞死亡的形式,其特征是铁依赖性的ROS积累[83],过度消耗NADPH使细胞对铁死亡敏感,而维持一定水平的NADPH可以促进细胞存活[84]。G6PD可以通过调节细胞内NADPH水平,调控铁死亡。这些研究表明,G6PD表达或活性的降低更容易导致肿瘤细胞死亡。肿瘤中G6PD的调控及作用见图2。
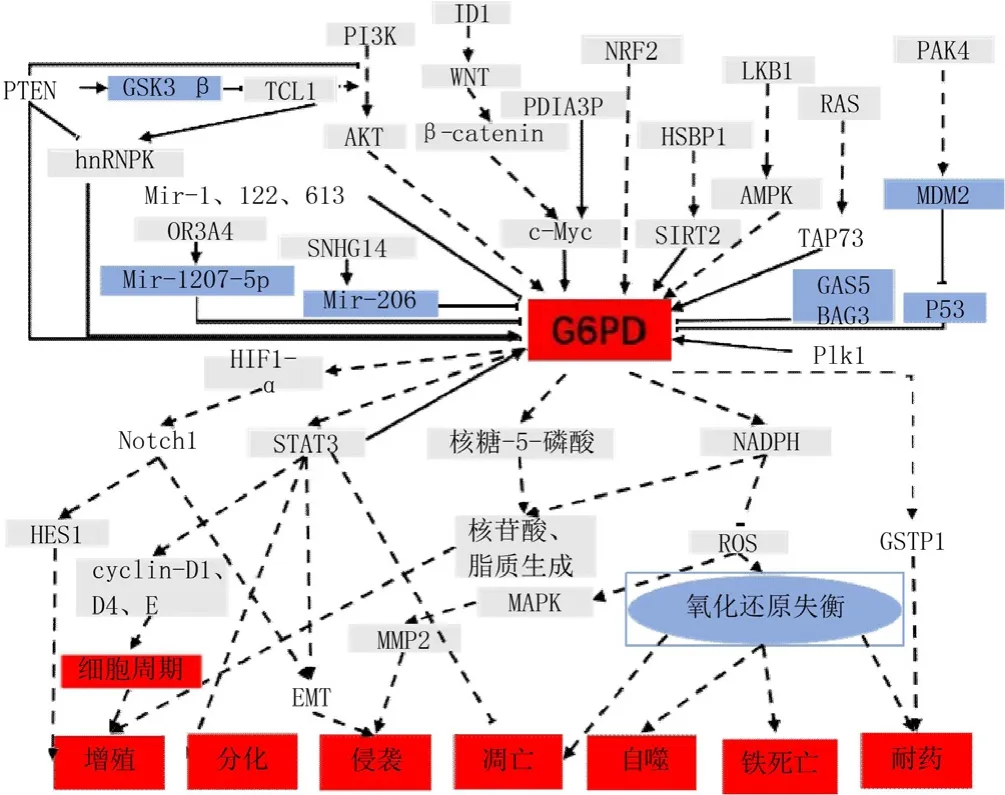
图2 肿瘤中G6PD的调控及作用Figure 2 Regulation and function of G6PD in tumors
3 G6PD可作为潜在的抗肿瘤治疗靶点
G6PD为肿瘤细胞提供NADPH以维持细胞内氧化还原平衡和脂类合成,并为核苷酸的生成提供R-5-P,因此在肿瘤的增殖、分化、凋亡中起到了重要作用。由于G6PD激活的肿瘤中NADPH水平较高,可以使这些肿瘤细胞免受氧化应激的影响,并对传统的抗肿瘤药物产生耐药。所以,在具有高度侵袭性和耐药性的肿瘤中经常检测到G6PD的高表达水平或活性升高。
G6PD能否成为抗肿瘤治疗的靶点?上述内容表明,抑制G6PD将限制肿瘤细胞的核酸、脂质等物质的合成及抗氧化能力,从而抑制肿瘤的进展、转移。目前,实验中使用的G6PD抑制剂包括NADP+类似物,如6-氨基烟酰胺(6-AN)、脱氢表雄酮(DHEA)等。体外试验中,6-AN的应用可以增加多种肿瘤细胞对顺铂和阿霉素的敏感性[66,85-86]。DHEA通过抑制G6PD活性,可降低甲基亚硝胺诱导的大鼠乳腺癌和前列腺癌的发生,以及辐射诱发的乳腺癌[87-89]。这些临床前研究结果显示了靶向G6PD在肿瘤治疗中的潜力。然而,DHEA在体内会转化为雄激素[90],这一效应极大地限制了DHEA抗肿瘤治疗的临床应用。另一种G6PD抑制剂6-AN也具有神经毒性。因此,开发具有成药性的G6PD抑制剂具有重要的临床研究价值。在天然产物中,表没食子儿茶素没食子酸酯(EGCG)、虎杖苷已被发现是G6PD的抑制剂[91-92],可能会在未来应用于临床。目前,新型靶向调节G6PD活性的抑制剂正在研发中。
ROS累积会对细胞内的DNA、脂类和蛋白质等大分子物质造成损害。虽然ROS能诱使肿瘤发生,但肿瘤细胞也必须抑制ROS水平,以避免高ROS水平对细胞内大分子物质的破坏,否则可能造成细胞死亡。一些利用氧化应激发挥抗肿瘤作用的药物,也可以通过调节G6PD从而影响抗肿瘤治疗的效果。这些药物通过升高NADPH水平使细胞免受氧化应激诱导的细胞凋亡[93]。耐药的P388小鼠淋巴细胞白血病模型中的G6PD活性比P388细胞高40%[94]。同样,耐阿霉素的结肠癌细胞由于G6PD活性较高而导致药物积累减少[86]。这些研究说明了G6PD介导的高水平NADPH在肿瘤的耐药中起着关键作用。同样,NADPH水平也与肿瘤的放疗敏感性相关。G6PD活性增加可以使细胞免受辐射损害[95]。6-AN通过G6PD抑制DNA的修复,同时增加ROS水平,诱使头颈部鳞状细胞癌和神经胶质瘤细胞的辐射损伤加剧[96]。其原理是G6PD抑制剂的应用使ROS水平升高,导致抗氧化防御受损,从而使肿瘤细胞选择性的放/化疗增敏。以上研究表明G6PD在调节化疗和放疗诱导的细胞死亡中起关键作用。因此,靶向G6PD可能为抗肿瘤治疗提供新的选择。
4 总结
根据肿瘤的异常能量及代谢改变状况来判断,作为葡萄糖代谢的重要分支,PPP水平的升高也是肿瘤区别于正常细胞的特征之一。肿瘤细胞通过PPP生成的R-5-P和NADPH以满足其快速增殖的核酸、脂类等物质的合成需求,同时大量生成的NADPH可用于维持细胞内的氧化还原平衡。PPP的增强也可能使基于氧化应激以及DNA损伤的抗肿瘤治疗效果减弱。G6PD作为PPP的关键酶,为临床通过G6PD实施的抗肿瘤治疗手段提供了理论依据。靶向G6PD可以抑制肿瘤细胞的快速发展,也可以通过联合用药的方式提高肿瘤细胞对其他化疗药物及放疗的敏感性。因此,开发成药性的G6PD抑制剂具有重要的临床价值。此外,PPP氧化分支的其他关键酶以及非氧化分支的限速酶也与肿瘤的发生相关,其所参与的细胞进程以及调控这些酶的上游信号通路同样需要深入研究。
作者贡献:孙键、陈艳、黄佩进行文章的构思与设计;孙键进行研究的实施与可行性分析;孙键、毛星星、龙圆圆进行资料收集;孙键、沙梦琪、刘喜平进行资料整理;孙键、黄佩撰写论文;孙键、陈艳、黄佩进行论文的修订;陈艳、黄佩负责文章的质量控制及审校,对文章整体负责,监督管理。
本文无利益冲突。
本文检索策略:
以“葡萄糖-6-磷酸脱氢酶(G6PD)和肿瘤”为关键词,检索PubMed数据库自建库至2020-11-30的相关文献。纳入标准:(1)G6PD在肿瘤中的调控及作用的研究;(2)G6PD在肿瘤的发生和治疗方面的最新研究进展。排除标准:重复及与主题不相关的文献。根据纳入与排除标准,最终纳入相关文献96篇。



