陈玉璞,张 毅,董智攀
(焦作市食品药品检验所,河南焦作 454000)
氨咖黄敏胶囊为感冒类非处方药品,适用于缓解普通感冒及流行性感冒引起的发热、头痛、四肢酸痛、流鼻涕、鼻塞、咽痛等疾病,临床应用广泛,生产厂家众多。本实验建立同时测定对乙酰氨基酚和马来酸氯苯那敏两种成分含量的高效液相色谱法,为控制药品质量,提供了一种简单、灵敏、快速准确的检测方法。
1 仪器与试药
1.1 仪器
UVIKONXL型紫外分光光度计;AE-240双量程电子天平(瑞士梅特勒公司);岛津高效液相色谱仪(LC-10ADVP泵、SPD-10AVP紫外检测器、CS-Light Potrun Analysis数据处理系统)。
1.2 试药
对乙酰氨基酚对照品(中国药品生物制品检定所,批号:100018-200408,供HPLC含量测定、比色检查用);马来酸氯苯那敏对照品 (中国药品生物制品检定所,批号:100047-200606,供含量测定用);氨咖黄敏胶囊(河南天工药业有限公司,批号:090505;河南兴源制药有限公司,批号:0903390;郑州康立制药有限公司,批号:090501);三乙胺为分析纯,水为纯净水,甲醇为一级色谱纯。
2 方法与结果
2.1 色谱条件
色谱柱:Shim-Paok CLC ODS C18(6.0 mm×200 mm,5 μm)色谱柱;流动相:甲醇-0.05 mol/L磷酸二氢钾溶液-三乙胺(10∶90∶0.02)(用磷酸调节 pH 至 3.4); 检测波长:215 nm;流速:1.0 ml/min;柱温:30℃;进样量:20 μl。
2.2 检测波长的选择
取对照品溶液,在180~300 nm的波长范围内进行扫描。结果在215 nm的波长处有最大吸收,故选用215 nm为检测波长。
2.3 对照品溶液的制备
取105℃干燥3 h的马来酸氯苯那敏对照品0.026 0 g,置100 ml量瓶中,用流动相稀释至刻度,摇匀。另精密称取对乙酰氨基酚对照品0.115 1 g,置50 ml量瓶中,同时精密加入马来酸氯苯那敏对照品溶液2 ml,用流动相稀释至刻度,摇匀,即得。
2.4 供试品溶液的制备
取本品10粒,除去胶囊后,精密称取细粉适量(约相当于对乙酰氨基酚120 mg),置50 ml量瓶中,用流动相充分溶解,并稀释至刻度,摇匀,用0.45 μm的微孔滤膜滤过,取续滤液即得。
2.5 阴性对照试验
按氨咖黄敏胶囊的处方工艺,不加对乙酰氨基酚、马来酸氯苯那敏,制成阴性对照品,同“2.4”项下方法制成阴性溶液。在选定的色谱条件下,进样20 μl,从色谱上看出,在该色谱条件下,在保留时间(tR)4.6 min与 18.9 min处,供试品和对照品溶液中出现两个色谱峰,而阴性对照溶液未见此色谱峰,表明在本实验条件下,对乙酰氨基酚、马来酸氯苯那敏的测定不受其他杂质峰干扰。见图1。
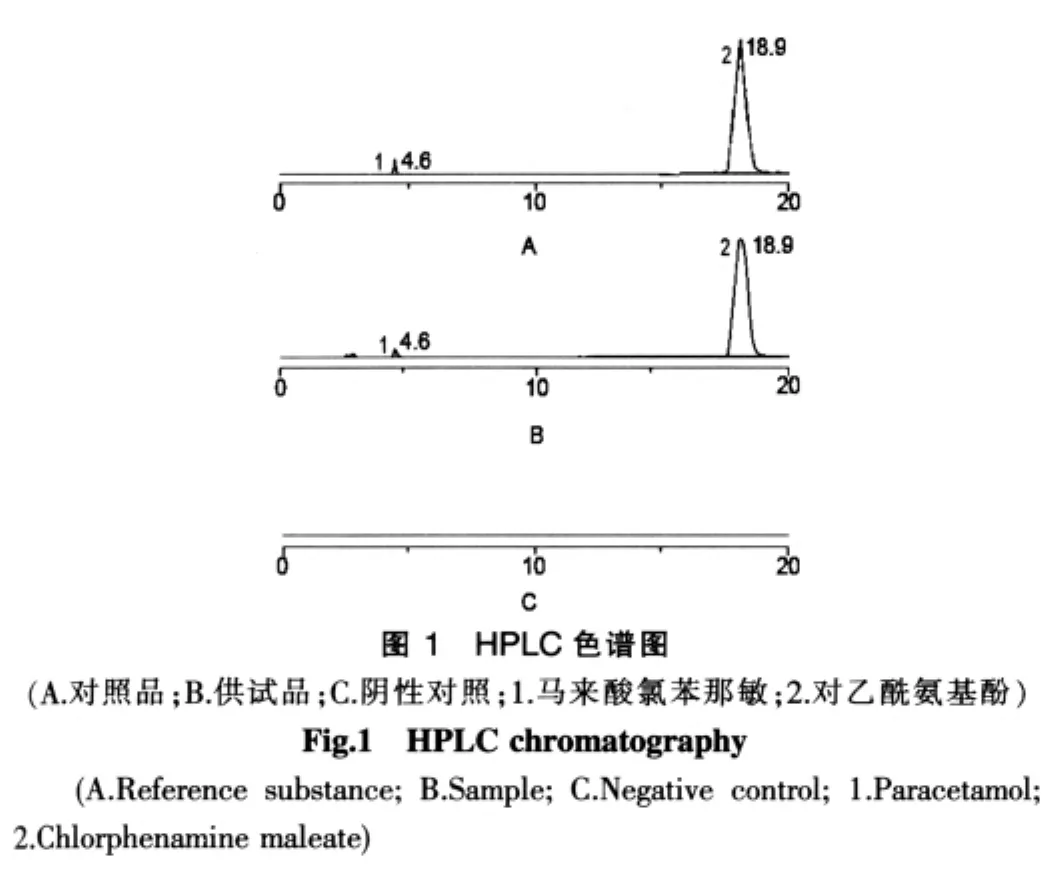
2.6 线性关系考察
精密量取对照品溶液 5.0、10.0、15.0、20.0、25.0 ml分别置于25 ml量瓶中,加流动相稀释至刻度,摇匀,即得。分别准确进样20 μl,按色谱条件测定其峰面积,以对照品进样量(μg)为横坐标,峰面积为纵坐标,计算回归方程,结果表明对乙酰氨基酚在0.092~4.604 μg范围内,呈良好的线性关系。回归方程为 Y=40 999.75X+2 258 190(r=0.999 8);马来酸氯苯那敏在4.160~20.800 μg范围内,呈良好的线性关系。回归方程为 Y=50 007.36X+89.20(r=0.999 7)。
2.7 精密度试验
取浓度为0.92 mg/ml的对乙酰氨基酚对照品与浓度为4.16 μg/ml的马来酸氯苯那敏对照品混合溶液重复进样5次,分别测定峰面积,结果显示,对乙酰氨基酚RSD=0.83%,马来酸氯苯那敏RSD=0.62%,结果表明该法精密度良好。
2.8 稳定性试验
取精密度试验项下的对照品溶液每间隔1 h进样1次,共8次,结果对乙酰氨基酚RSD=1.57%,马来酸氯苯那敏RSD=1.72%。结果表明样品在8 h内基本稳定。
2.9 重复性试验
取样品(批号:090505,河南省天工药业有限公司)适量,按“2.4”项下方法制备样品溶液,依法平行测定6次,结果对乙酰氨基酚RSD=1.62%;马来酸氯苯那敏RSD=1.67%。结果表明该法的重现性较好。
2.10 加样回收率试验
取样品 (批号:090505,河南省天工药业有限公司)约0.14 g,共6份,精密称定,加入对乙酰氨基酚对照品36.8 mg、马来酸氯苯那敏对照品166.4 μg,按“2.4”项下方法制备供试品溶液,按色谱条件测定,结果见表1、2。
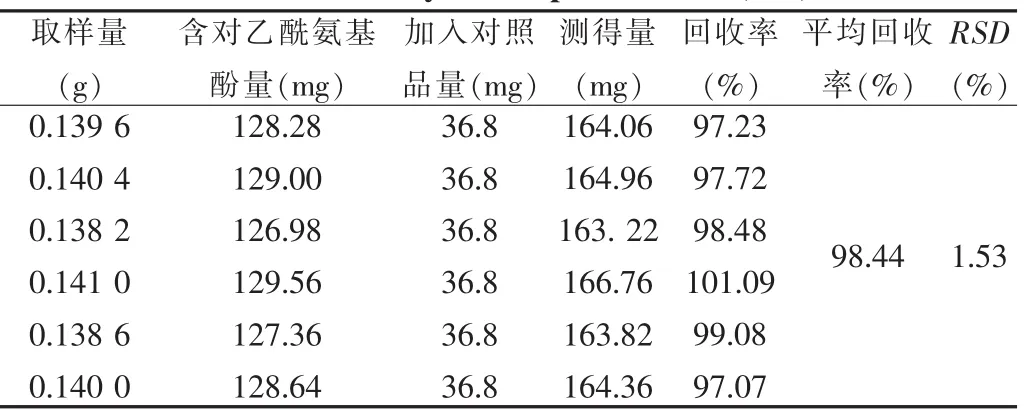
表1 对乙酰胺基酚加样回收率(n=6)Tab.1 Recovery test of paracetamol(n=6)
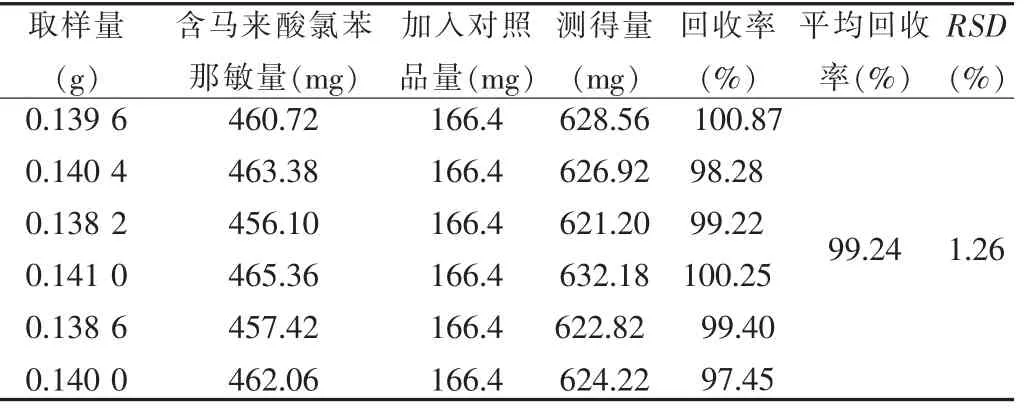
表2 马来酸氯苯那敏加样回收率(n=6)Tab.2 Recovery test of chlorphenamine maleate(n=6)
2.11 样品含量测定
取3批样品各10粒,除去胶囊后,各精取细粉适量(约相当于对乙酰氨基酚120 mg),分别置于50 ml量瓶中,用流动相溶解,并稀释至刻度,摇匀,用0.45 μm的微孔滤膜滤过,取续滤液作为供试品溶液;另取对乙酰氨基酚、马来酸氯苯那敏对照品适量,用流动相制成每毫升中含对乙酰氨基酚1.15 mg/ml和马来酸氯苯那敏5.20 μg/ml的溶液。分别进样20 μl,按上述色谱条件,测定色谱峰面积,结果见表3。
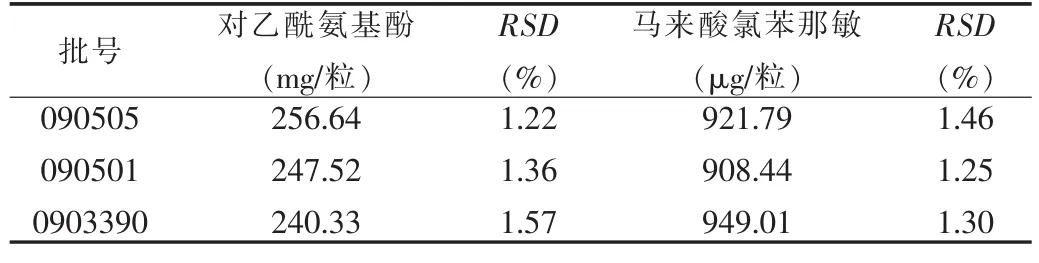
表3 样品含量测定结果Tab.3 Content determination results of samples
3 讨论
氨咖黄敏胶囊,临床应用广泛,生产厂家众多,而《国家药品标准化学药品地方标准上升国家标准第三册》中没有马来酸氯苯那敏的含量测定方法,无法保证药品质量;而对乙酰氨基酚的检测方法为亚硝酸钠滴定法,终点不易控制,常常误差偏大。本实验经过对多批次样品中马来酸氯苯那敏、对乙酰氨基酚的含量测定表明,该方法简单、灵敏、准确,可以控制产品的质量。
从氨咖黄敏胶囊的配方可以看出,对乙酰氨基酚含量远远高出马来酸氯苯那敏的含量,所以进样量浓度一定要控制好。若浓度过高,则对乙酰氨基酚的峰形易出现平头峰,无法计算,若浓度过低,则马来酸氯苯那敏不易检测到,且误差偏大。
[1]国家药典委员会.中华人民共和国药典[S].一部.北京:化学工业出版社,2005:附录Ⅴ D:28-30.
[2]范剑,米振清.氨咖黄敏胶囊中对乙酰氨基酚、咖啡因含量的测量不确定度评定[J].中国医药导报,2009,6(11):52-53.
[3]刘雅静,徐亚杰,孙苓苓.一阶导数一多元线性回归法同时测定氨咖黄敏胶囊三组分的含量[J].山西医药杂志,2009,38(6):538.
[4]国家药典委员会.化学药品地方标准上升国家标准第三册[S].北京:化学工业出版社,2002:50-52,263-266.
[5]赵素珍,宋卫云.高效液相色谱法测定清热止泻合剂中葛根素的含量[J].中国医药导报,2007,4(30):87-88.
[6]周岐勋,李健和,徐幸民.高效液相色谱法测定盐酸甲氯芬酯分散片含量及有关物质[J].中国当代医药,2009,16(4):73-76.



