柳中辉 王家明 张黎明 孙华
天津科技大学生物工程学院工业发酵微生物教育部重点实验室,天津 300457
微粉化萘丁美酮的工业制粒工艺研究
柳中辉 王家明 张黎明 孙华
天津科技大学生物工程学院工业发酵微生物教育部重点实验室,天津 300457
目的通过优化制粒溶液量百分比和制粒时间,将微粉化的萘丁美酮制成适合片剂生产的颗粒。方法以微粉化的萘丁美酮作为主要活性成分,以萘丁美酮超过80%(W/W)处方配比的条件下,以纯水、羟丙基甲基纤维素E-3和十二烷基硫酸钠为制粒溶液,将萘丁美酮,羟基乙酸淀粉钠和微晶纤维素PH101进行制粒,流化床干燥,并对制得的颗粒进行物理性状检测,包括颗粒外观、颗粒粒径分布、颗粒堆实密度及可压性指标。然后将颗粒压片,检测片剂的硬度、崩解度及和溶出度,获得制粒溶液量百分比和制粒时间的最优值。结果制粒溶液量30%(W/W)和制粒时间60 s时,为最佳制粒工艺参数。结论以此工艺进行制粒,可达到适合工业生产的片剂指标。
微粉化;萘丁美酮;制粒工艺;片剂
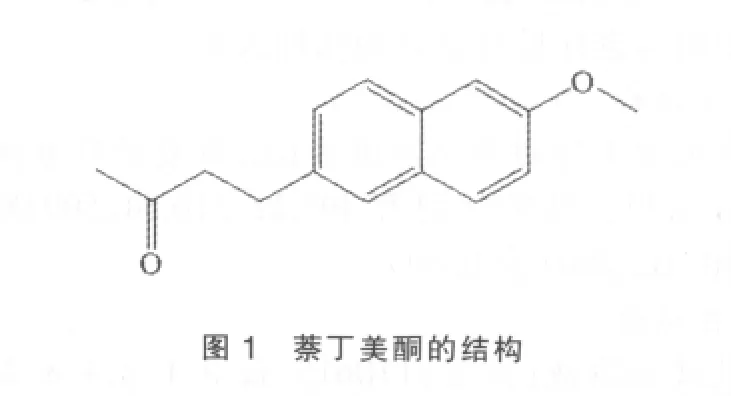
萘丁美酮(Nabumeton)又称萘普酮、瑞力芬、萘布美通、纳布美通、萘美酮。其化学名为4-(6-甲氧基-2-萘基)-2-丁酮(图1),该物质为白色结晶粉末,属于酮型结构的前体药物,在肝脏内可被代谢为6-甲氧基-2-萘乙酸,活性代谢物无肝肠循环,几乎全部经尿液排出体外,胃肠道反应小、副作用少、耐受性好,具有抗炎、止痛和解热作用。主要用于风湿性、类风湿性关节炎、骨关节炎[1]、软组织损伤、强直性脊椎炎,对前列腺素合成酶也有抑制作用[2]。萘丁美酮是英国Beecham公司开发的非甾体类抗炎药,于1985年在爱尔兰上市。萘丁美酮的水溶性很差,可通过改变其晶型提高溶解度[3],也可进行微粉化处理改善颗粒的润湿性,以提高水不溶性药物的溶解度和溶解速率[4]。微粉化是以先进的物理或化学的手段将物料制备成微米级或以下粉体的过程,其操作简单技术要求不高,工业上广泛应用此方法提高原料的溶解性[5]。但微粉化的弊端是增大了物料颗粒的表面积,流动性降低,在固体制剂生产中,物料的流动性对产品质量有重要影响[6]。本研究在工业化规模的制粒设备上,通过正交实验设计对影响颗粒及压片的2个关键工艺参数(制粒溶液量和制粒时间)进行考察,以期寻找改善流动性差的微粉化萘丁美酮颗粒的重要影响参数,同时保证压片的结果符合质量标准。
1 仪器与材料
高速剪切制粒机(德国DIONSA P25),Glatt-50流化床(德国Glatt-50),水份测定仪HB43-S(瑞士梅特勒HB43-S),振实密度仪(成都精新粉体测试设备公司JZ-7),高速压片机(德国Fette 2000),光学显微镜(德国Nano Science Standard Zeta-20),X激光粒度分析仪(英国马儿文MS2000)。百分之一分析天平(瑞士梅特勒AB-L)。
萘丁美酮(重庆西南合成制药厂,含量>99.0%,批号:10010479),十二烷基硫酸钠(美国威特科公司),羟基乙酸淀粉钠(法国罗盖特公司),羟丙基甲基纤维素E-3(海卡乐康包衣技术有限公司),微晶纤维素PH101(美国富美实FMC公司)。
2 方法与结果
2.1 微粉化萘丁美酮的制粒工艺
用纯水将羟丙基甲基纤维素E-3和十二烷基硫酸钠溶解至透明的制粒溶液,将萘丁美酮,羟基乙酸淀粉钠和微晶纤维素PH101投入到高速剪切制粒机中,低速搅拌时加入上述制粒溶液,然后高速搅拌进行制粒,当达到目标电流值关闭制粒机,将湿颗粒投入到流化床进行干燥,达到目标LOD值后,对制得的颗粒进行物理性状检测,包括颗粒外观、颗粒粒径分布、颗粒堆实密度及可压性指标(Carr′s)。然后用高速压片机对颗粒进行压片即得萘丁美酮片剂,并对片剂进行硬度、崩解度及溶出度检测,选择最优的制粒溶液量百分比和制粒时间。
2.2 实验设计
以制粒溶液百分比、制粒时间为实验因素,根据预实验条件确定2种因素的2种不同水平,采用L4(22)安排正交实验表,实验设计见表1。

表1 因素和水平
2.3 实验结果
2.3.1 颗粒外观检测
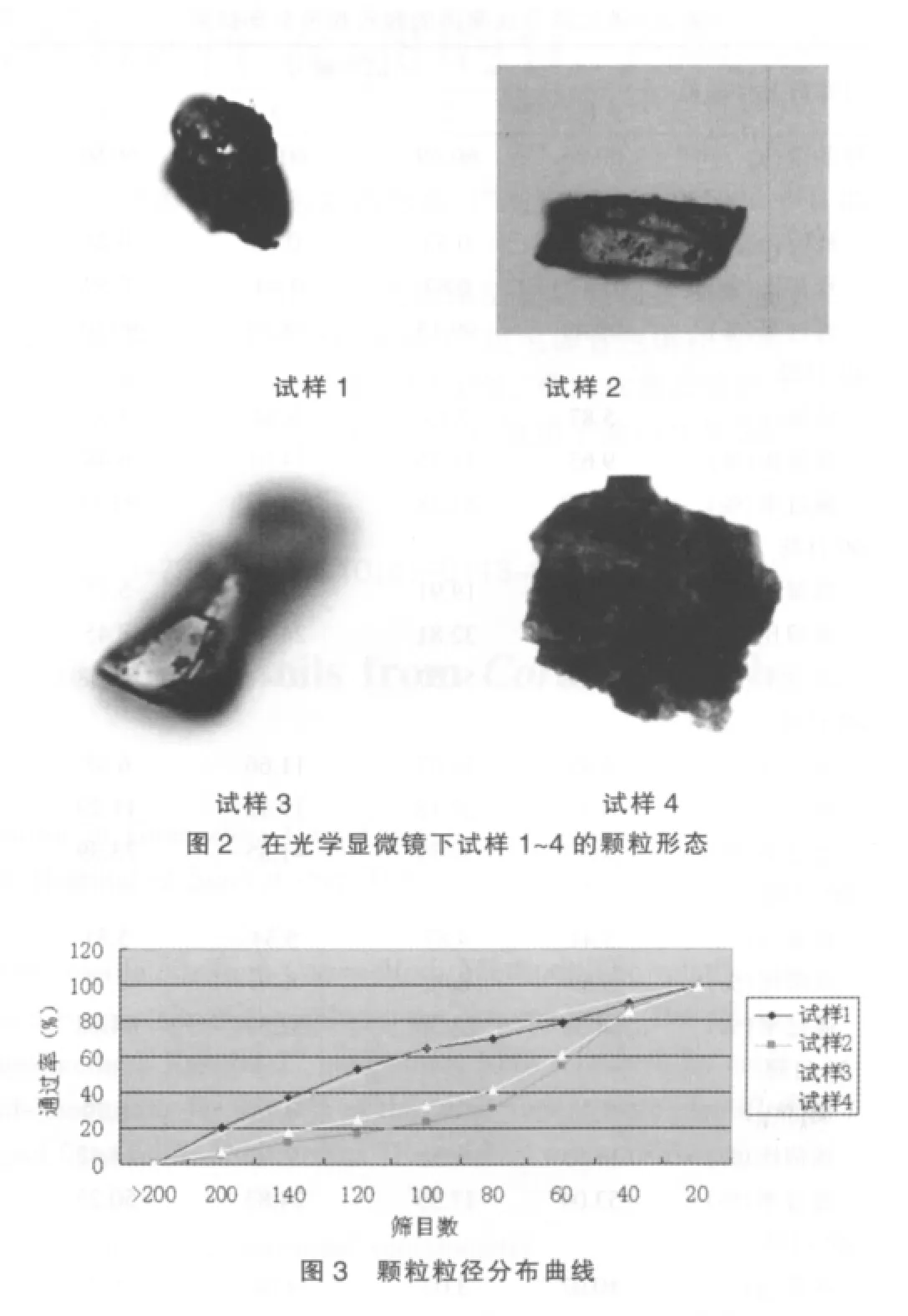
用光学显微镜法可以观察颗粒的真实形态和大小。用压缩空气将颗粒分散在玻璃样品座上,用光学显微镜自带的相机对每个颗粒进行拍照,之后通过软件对颗粒的大小数据进行分析。见图2。
在显微镜下观察试样1~4的颗粒形态,由图2可见,试样1和3的样品颗粒圆整度较好,而试样2和4颗粒呈现出明显的棱角。因此,制粒时间长,颗粒受到的剪切力较多,容易破坏颗粒的圆整度。
2.3.2 颗粒粒径分布检测
称取经流化床干燥的颗粒,每个试样各取不少于50 g样品。将标准筛按照孔径由大至小的顺序叠好,并装上筛底,将称好的试样倒入最上层筛板,加上筛盖,安装在振荡机上。振荡机震动3min后,取下筛板。分别称量各筛上和底盘中试样的质量,见表2。
通过对表2中颗粒粒径分部的数据作图,得到粒径分布图(图3)。
从筛分法所得到的颗粒数据可以看出,试样1和4的颗粒分布相接近,而试样2和3的颗粒分布相接近。试样2和3的颗粒中值(D50)大于试样1和4。因此,制粒溶液的量和颗粒大小有关,并与D50呈正向线性关系。
2.3.3 颗粒堆实密度及可压性指标(Ca r r′s)检测
2.3.3.1 松密度的测定操作前取一洁净、干燥的量筒(100mL),并称其质量(m0,g)。将通过20目筛的样品松缓地转入量筒中至(90±5)mL处,称量量筒与样品的质量(m1,g),精确到0.1 g,抚平粉末表面,读取固体粉末的体积(V1,mL)。计算公式:松密度=(m1-m0)/V1×100。
2.3.3.2 紧密度的测定将上述盛有样品的量筒放在台面上(铺有约5mm厚的橡胶),由2 cm左右的高度自坠到台面上,反复此操作约100次,量得压紧后的粉末体积(V0,mL),继续上述操作约30次,量得粉末体积(V2,mL)。当V0与V2相差小于2 mL时,读取终体积(V2,mL),否则重复上述操作,直到符合为止。计算公式:紧密度=(m1-m0)/V2×100。见表3。
通过可压性指数Carr′s值分析得知,试样2和3所得颗粒的Carr's指数较低,体现出较好的流动性,而试样1和4的颗粒Carr′s指数较高,流动性下降。说明制粒时间及制粒溶液的量对颗粒的可压性指数成反比关系,与流动性成正比关系。因此,实际生产中可通过增加制粒时间和提高制粒溶液的用量来提高颗粒的流动性。
2.3.4 压片硬度检测
在Fette2000高速旋转压片机上对4个试样颗粒进行压片,开机设定理论片重为610 mg,单片的片重限度为±3%(592~628mg/片),平均片重限度±1%(604~616mg/片),调整压片机主压力轮和预压力,保证片子厚度在5.95~6.05 mm间,开始压片并记录片重及片子硬度结果,片子硬度的范围为69~140 N。见表4。
从素片硬度结果可以看到试样1颗粒所制得的片剂硬度明显偏低,部分样品已经超过低限值(<69 N),试样2颗粒所得片剂的硬度明显偏高,也有几个点超过上限(>140 N),而试样3和4的硬度较为适中。通过数据可以发现,制粒时间和制粒溶液量同时增加,片剂的硬度偏高。
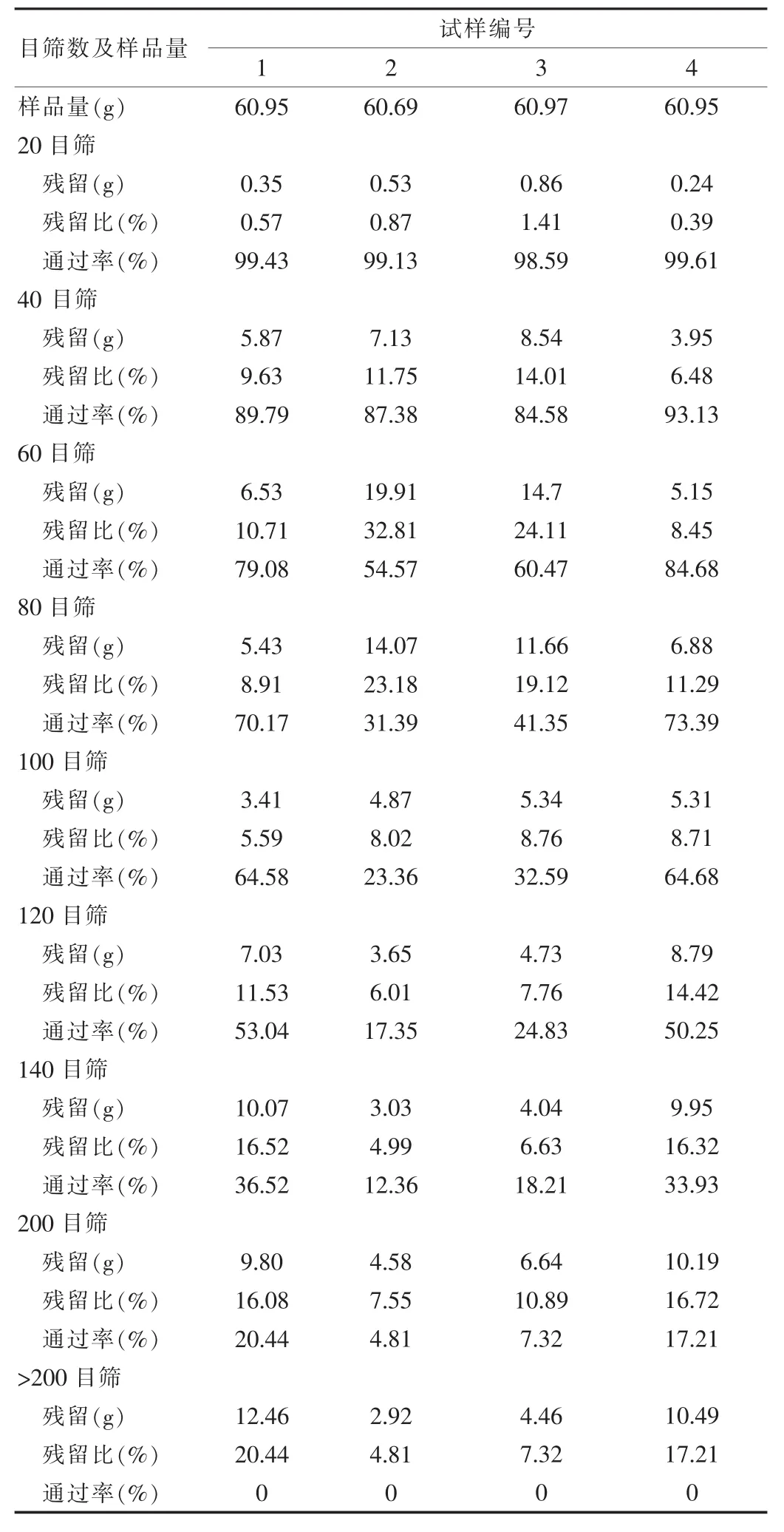
表2 通过筛分法测得的颗粒粒径分部结果
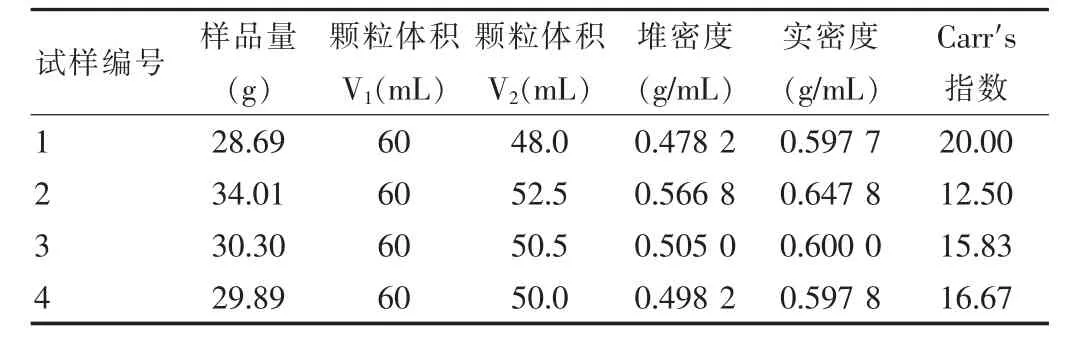
表3 颗粒堆实密度及Carr′s结果
2.3.5 片子崩解及溶出度检测
通过崩解仪及溶出仪对所得片剂进行崩解和溶出度测定,标准的崩解时限为15min内崩解完全,溶出度的标准为不得少于标识量的85%。见表5。

表4 压片片重及硬度结果
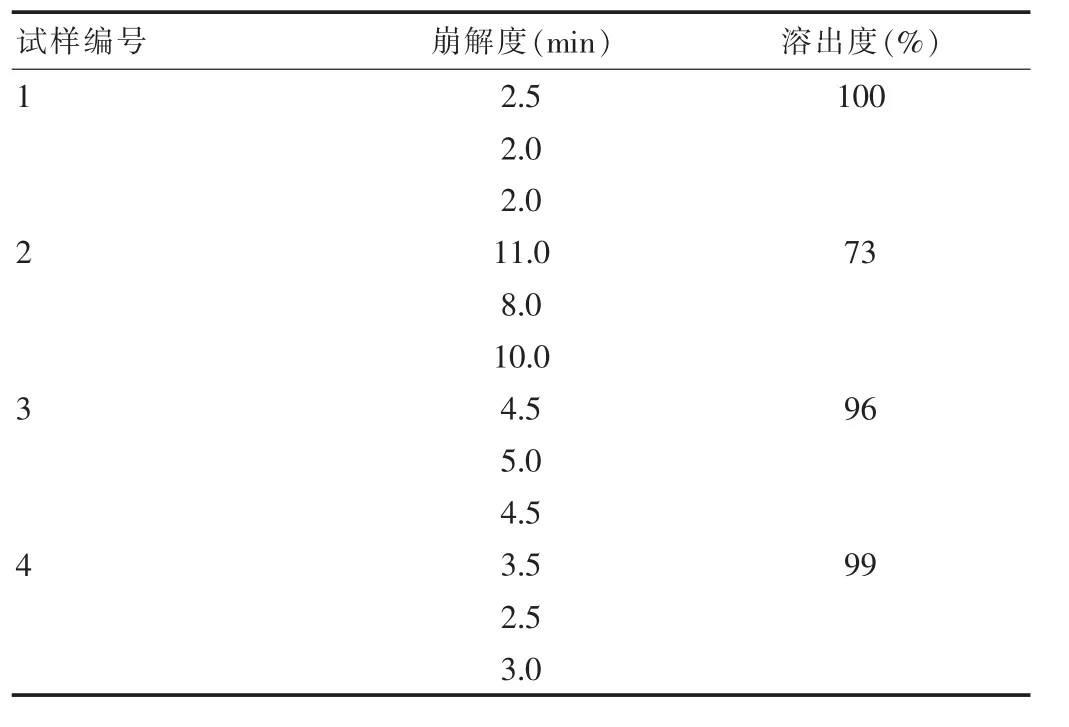
表5 崩解及溶出度结果
从素片崩解结果可以看出,试样1颗粒所制得的片剂的崩解时间最短,溶出度也较好。而试样2的片子的崩解时间明显延长,溶出度的结果也未达到设定标准,而试样3和4的崩解和溶出度均符合要求。这说明制粒时间越长,制粒溶液加入量越多,片剂越难溶解。
3 讨论
本研究通过正交实验设计对影响颗粒及压片的2个关键工艺参数制粒溶液和制粒时间进行考察,通过对颗粒外观、颗粒粒径分布、颗粒堆实密度、可压性指标以及片剂的硬度、崩解度、溶出度多项实验结果进行比较得知,试样3的制粒时间(60 s)和制粒溶液量(30%,W/W)为最佳工艺条件,即可改善微粉萘丁美酮颗粒的流动性同时也可以保证最终压片结果符合质量标准。
[1]陈雅,张彦.奈丁美酮搽剂的制备及质量控制[J].军队医药,2010,9(4):31-32.
[2]陈小全,左之利.奈丁美酮合成的改进[J].有机化学,2010,30(7):1069-1071.
[3]Purushotham R,Kamamia EK.Characterisation of Nabumetone Microcrystals Prepared by Solvent Evaporation Method[J].International Journal of Research in Pharmaceutical and Biomedical Sciences,2011,2(3):1346-1349.
[4]陈鹏,张小岗.微粉化技术提高水不溶性药物溶解度[J].化学通报,2007,70(10):766-771。
[5]徐月红,王宁生.中药微粉化的现状与分析[J].中国中药杂志,2004,29(6):497-500.
[6]崔福德.药剂学[M].5版.北京:人民卫生出版社,2003:298.
Study ofm icronized Nabumetone industrial granulating process
LIU Zhonghui WANG Jiaming ZHANG Liming SUN Hua
Key Laboratory of Industrial Microbiology of Ministry of Education College of Biotechnology,Tianjin University of Science and Technology,Tianjin 300457,China
ObjectiveTo determine suitable Nabumetone granule for tablet production through optimizing the percentage of granulation solution and granulation time.MethodsMicronized Nabumetone was used as an active pharmaceutical ingredient(APIs),with the prescription matching conditions of Nabumetone of more than 80%(W/W),blending with Sodium Starch Glycolate and avicel PH10,and then adding the granulation solution which mixed with purified water,HPMC E-3 and sodium lauryl sulphate.The granulewas gotten after drying in fluid-bed dryer.Sample was taken from dried granule to perform physical property testing which includes granule appearance,particle size distribution,bulk type density,conducting compression with the granule and testing the tablet hardness,disintegration and dissolution in order to optimize granulation parameters.ResultsThe optimized parameters had been identified,which were 30%(W/W)granulation solution and with granulation time of 60 s,respectively.ConclusionGood tablet quality attributes can be achieved with these granulation parameters.
Micronized;Nabumetone;Granulating process;Tablet
R284.2
A
1673-7210(2012)10(c)-0110-04
天津市应用基础及前沿技术研究计划项目(项目编号:11JGYBJC14300)。
孙华(1977.10-),女,博士研究生,副教授,硕士生导师,主要从事新药研发工作。
2012-05-22 本文编辑:卫轲)



