邵滢霏 董冰子
(1 温州医科大学仁济学院,浙江 温州 325035;2 青岛大学附属医院内分泌科)
巴尔得-别德尔综合征(Bardet-Biedl syndro-me,BBS)是一种常染色体隐性遗传性疾病,患者的特点是向心性肥胖、心智损害、视锥-视杆营养不良、轴后性多指、性腺机能减退以及肾脏异常,该病目前至少已经确定了19个致病基因。目前国内报道基因确诊的BBS病例约40例,但尚未见存在BBS5基因变异致病的报道[1-2]。另外,肾脏异常和终末期肾功能衰竭是BBS患者的主要死亡原因,存在于绝大多数患者中,但在5型BBS患者中罕见[3]。本研究对1例临床表现为严重肾功能损害的5型BBS患者的表型和基因型进行分析,以期能够提高临床医生对该病的认识。现将结果报告如下。
1 临床资料
患者,男,31岁,汉族,系孕1产1,足月顺产,出生体质量4.2 kg。出生时发现双手六指畸形(轴后性)、双足六趾畸形(轴后性)。生后多饮、多尿、多食,体质量增长明显快于同龄儿童,但学语迟缓。1岁时行双手多指切除术。4岁时仍有尿床,7岁时发现视力低下伴夜盲,屈光矫正疗效不显着。20岁时在当地查体发现肾功能异常(血肌酐190 μmol/L)、血糖异常和血脂异常,未进一步系统治疗。患者于2021年10月就诊于青岛大学附属医院肾病科,体格检查:身高172 cm,体质量99.0 kg,体质量指数(BMI)33.5 kg/m2,血压156/90 mmHg,体型肥胖;前额窄,呈特殊面容,头发稀疏,双足六趾畸形,双手小指根部见1 cm左右手术瘢痕;心、肺、腹部检查均无明显异常,外生殖器发育正常,步态正常。实验室检查结果:血红蛋白90.0 g/L,血肌酐730 μmol/L,属慢性肾脏病5期,尿酸613 μmol/L,总胆固醇2.33 mmol/L,三酰甘油0.48 mmol/L,空腹血糖5.54 mmol/L,血钙2.39 mmol/L,甲状旁腺激素(PTH)1 345 ng/L,血磷2.21 mmol/L;尿液分析显示尿蛋白(+),尿糖(),尿微量白蛋白/肌酐比值为14 mg/g。肾脏CT显示双肾缩小伴囊肿样改变。眼科检查:右眼视力0.05,左眼视力0.12。因无中心注视无法进行视野检测。右眼黄斑区见椭圆形萎缩改变,边界清晰;左眼黄斑区病灶边界模糊,中心密集黄色斑点,双眼黄斑病灶周边呈色素样改变。眼底荧光造影示双眼黄斑透见荧光,周围环状色素荧光遮蔽。光相干断层成像术(OCT)显示双眼黄斑区视网膜全层萎缩,右眼和左眼的黄斑区视网膜厚度分别为130、125 μm,视网膜电图显示视锥和视杆细胞均有不同程度损害。临床诊断为终末期肾功能衰竭,BBS[视杆-视锥营养不良、肥胖、肾损害、多指(趾)、糖尿病、颅面异常],继发性甲状旁腺功能亢进。治疗方案:血液透析治疗;采用活性维生素D冲击治疗继发性甲状旁腺功能亢进,由于效果欠佳,择期给予了甲状旁腺全部切除联合部分前臂埋置,术后血液中钙-磷-PTH水平均逐渐恢复正常。患者透析至今,一般情况良好。
该患者父母否认近亲结婚。对先证者取外周血2 mL,提取基因组DNA并采用二代测序技术进行全外显子捕获和高通量测序,结果显示BBS5基因有2个杂合变异,分别为c.1A>G(p.Met1?)(NM_152384.3)和1~5号外显子的大片段缺失[2q31.1区域约8 500 bp杂合缺失c.(?-60)_(386+1_387-1)del]。采集先证者父母外周血2 mL,通过Sanger测序进行致病基因位点的验证,结果证实c.1A>G(p.Met1?)来自其母亲(图1);基于二代测序技术进行的大片段拷贝数变异分析证实,1~5号外显子的大片段缺失[c.(?-60)_(386+1_387-1)del]来自其父亲。根据2015年美国遗传学会(ACMG)遗传变异分类标准与指南进行分析,2个变异均为致病性变异。另外,发现患者携带PKD1新的基因错义变异c.9331T>A(p.F3111I)(NM_001009944.3,dbSNP rs767845814),该突变来自其母亲;经在线软件MutationTaster和PolyPhen-2预测该变异有害,但PROVEAN预测为中性;该变异在GnomAD的最小等位基因频率为0.000 004,该密码子位点未见其他变异报道;根据2015年ACMG,评估为意义未明变异。
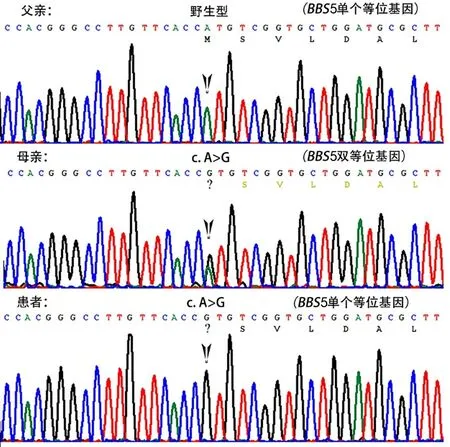
图1 该家系BBS5突变位点c.1A>G Sanger测序验证图Fig.1 Sanger sequencing validation of the c.1A>G mutation site of BBS5 in the family
2 讨 论
不同地区BBS的患病率有所不同,但目前仍缺乏亚洲人群的患病率数据。MULLER等[4]发现5型BBS仅占BBS(主要来自高加索人)的2%左右。目前全球共报道5型BBS家系21个,发现变异基因22个,而且报道主要来自中东地区[4-15],而来自亚洲仅2个家系(日本和菲律宾各1个)[5-6]。中国目前未见确切报道。申金凤等[16]报道了1例疑似5型BBS的患儿,但其BBS的诊断依据缺乏主要临床特征和次要临床特征,而且作为常染色体隐性遗传性疾病,仅仅发现1个杂合变异c.409T>C(p.Y137H),不能通过基因诊断确诊。刘兵等[17]针对1例BBS家系进行了基因连锁分析,结果证实发病与BBS5基因连锁有关,但由于没有进行BBS5基因测序分析,故不能完全确诊。本研究中患者为国内目前唯一通过基因检测确诊的5型BBS病例。
BBS是纤毛病的一种。纤毛是指凸起于真核细胞基体并沿微管轴丝向细胞表面外延的披膜细胞器。纤毛作为细胞信号天线,其在细胞分裂、器官形成和发育分化等生命过程中不可或缺。纤毛组装缺陷/信号传导功能障碍均可导致多种“纤毛病”。纤毛组装及其跨膜受体纤毛内稳态维持均依赖双向纤毛内运输(IFT)系统。在双向IFT的运输过程中,BBSome通过与IFT偶联来充当IFT与纤毛跨膜信号蛋白之间的衔接子,使得纤毛跨膜信号蛋白得以依赖IFT系统维持其纤毛内稳态。BBSome是由BBS1、BBS2、BBS4、BBS5、BBS7、BBS8、BBS9以及BBS18组成的8聚体蛋白复合物,目前发现任何一个亚基双等位基因突变均可导致BBS。
研究发现,绝大多数5型BBS患者为BBS5基因发生的变异,如无义变异、错义突变、起始密码子变异、编码框位移变异或外显子大片段缺失,其中错义变异仅占27%(6/22),且大多数为纯合变异,来自近亲结婚家庭。另外,已报道的6个错义变异中,p.L50R、p.T53I、p.R56G、p.G72S以及p.T183A分别位于BBS5编码蛋白BBS5的两个保守PH结构域中,推测可能对BBSome组装以及纤毛形成有影响。本研究中,发现先证者BBS5基因有2个杂合变异,其中c.1A>G(p.Met1?)是起始密码子,推测该变异可产生新的起始密码子,或致蛋白翻译起始障碍。以往的研究报道还发现有5型BBS患者的其他2种形式(c.1A>T和c.2T>A)的BBS5基因起始密码子突变,而核苷酸的改变与本例患者不同[4,7-8]。本研究中的先证者携带的另1个杂合变异是c.(?_-60)_(386+1_387-1)del(外显子1~5号的缺失)大片段缺失,为首次发现的变异,预测该变异将导致BBS5基因不能表达。
BBS的临床诊断标准应该具备以下4项主要临床特征,或者3个主要特征联合2个及以上次要特征[3]。主要临床特征包括视杆-视锥营养不良、轴后性多指(趾)、向心性肥胖、学习困难、性功能下降(男)或者生殖器异常(女)和肾脏异常,次要特征包括学语迟缓、发育迟缓、行为异常、眼睛异常(斜视、白内障)、短指/并指、共济失调、肌张力过高、糖尿病、口腔颌面部异常、心血管异常、肝脏受累、颅面异常(短头畸形、前额窄、男性正面秃顶、眼裂短窄、鼻梁下陷、面中部后缩等)。既往报道的5型BBS患者主要临床表型为视杆-视锥营养不良、肥胖和多指(趾),仅有4例患者呈现多囊肾、慢性肾功能不全、孤立肾等肾脏异常[4-16]。本研究中患者肾脏表型极为严重,已进入终末期肾功能衰竭,考虑可能与以下原因有关:①既往报道的患者多于学龄期确诊BBS,而本例患者确诊时已经31岁。②本例患者携带有PKD1基因的1个错义变异(p.F3111I)。PKD1为常染色体显性遗传性多囊肾致病基因,该突变来自于其母亲,虽然其母亲无多囊肾改变,但不排除PKD1突变对该患者在5型BBS肾损害的基础上的二次打击作用。③其他基因的协同作用,特别是不能排除纤毛病相关基因的协同作用。另外,本例患者在终末期肾功能不全前期存在血糖和血脂异常,但入院时实验室检查血糖等生化指标却均正常,推测原因可能与肾衰竭致胰岛素清除减少、胰岛素敏感性增加、糖异生减弱及饮食减少有关。
关于BBS的治疗方案,GENUIS等[18]对1例BBS10基因突变的患者给予辅酶Q10、肉毒碱、复合维生素B、叶酸、维生素D、精氨酸、酪氨酸和5-羟基色氨酸治疗,疗效显着,但仅限于病例报道,无大规模临床循证证据。也有研究者利用雷帕霉素等化合物在斑马鱼BBS模型中取得了改善肾脏结构和肾功能的成效,但未在哺乳动物中获得证实。动物实验的结果提示,局部基因治疗(给小鼠模型注射BBS的腺病毒载体)对小鼠的视力具有保护作用,这一研究的临床转化应用也值得期待。但本例患者在诊断时已经出现严重肾功能不全和视力损害,目前尚无临床治疗特效方法,因此目前的治疗措施主要以对症治疗为主。
综合以上,本研究报道了1例5型BBS患者的临床特征和表型,表现为肥胖、多指(趾)、视杆-视锥营养不良及肾功能不全,特别是肾功能不全快速进展为终末期肾病的合并症较罕见,丰富了疾病临床特征。本例患者的2个BBS5基因突变为新发突变,丰富了人类BBS5基因突变谱和5型BBS临床表型,为临床医生对该病表型特征的认识提供参考。
利益冲突声明:所有作者声明不存在利益冲突。
ConflictsofInterest: All authors disclose no relevant conflicts of interest.
伦理批准和知情同意:本研究涉及的所有试验均已通过青岛大学附属医院医学伦理委员会的审核批准(2021临审字第009号)。所有试验过程均遵照2013年修订的《赫尔辛基宣言》的条例进行。受试对象或其亲属已经签署知情同意书。
EthicsApprovalandPatientConsent: All experimental protocols in this study were reviewed and approved by The Medical Ethics Committee of The Affiliated Hospital of Qingdao University (Approval Letter No. TEM 009), and all experimental protocols were carried out by following The 2013 Declaration of Helsinki. Consent letters have been signed by the research participants or their relatives.
作者贡献:董冰子参与了研究设计;邵滢霏参与了论文的写作和修改。所有作者均阅读并同意发表该论文。
Contributions: The study was designed byDONGBingzi. The manuscript was drafted and revised bySHAOYingfei. All the authors have read the last version of the paper and consented submission.



