况安香, 曾晓萍, 曹 盼1,, 梁光义***, 徐必学***
(1.贵州中医药大学, 贵州 贵阳 550025; 2.贵州医科大学 省部共建药用植物功效与利用国家重点实验室, 贵州 贵阳 550014; 3.贵州省中国科学院天然产物化学重点实验室, 贵州 贵阳 550014)
乙型肝炎(乙肝)是由乙肝病毒(hepatitis B virus,HBV)感染引起的、严重威胁人类健康的疾病之一[1],目前治疗乙肝的药物主要有以α-干扰素为代表的生物类免疫调节剂和恩替卡韦等为代表的核苷(酸)类药物[2-3],但这些药物均不能完全治愈乙肝[4-5],因此,研究开发具有新化学结构类型的乙肝治疗药物具有重要意义。马蹄金素[N-(N-苯甲酰基-L-苯丙氨酰基)-O-乙酰基-L-苯丙氨醇,MTS,图1]是本课题组从苗药马蹄金中发现的一个具有抗乙肝病毒活性的先导化合物[6-8],其衍生物Y101已经获得临床批件并完成了一期临床试验[9-11]。鉴于MTS为二肽基本母核,与现在临床上的核苷类抗乙肝药物的基本骨架完全不同,值得广泛深入探讨MTS基本母核上不同取代基对其抗乙肝病毒的影响。对生物分子用卤素(Cl、Br、I和F)修饰是药物化学研究的热点,约40%的卤化药物进入市场或临床前试验阶段,约25%的有机卤化药物进入市场,而34%的卤化药物仍处于研发阶段[12- 13],这表明卤素在药物研发中起着重要的作用,其中又以含氟或氯取代的应用更为广泛[14]。为探讨含氟或氯取代对MTS衍生物在抗HBV活性方面的影响,本研究设计合成了一系列含氟或氯取代的MTS衍生物,并对其进行了抗HBV的评价。
1 材料与方法
1.1 仪器、试剂
仪器有XT-4型熔点仪(温度未校正)、德国布鲁克公司AVANCE NEO-600 MHz型超导核磁共振仪、美国Varian公司Inova-400 MHz型超导核磁共振仪(以TMS为内标)、美国Hewlett-Packard公司HP-5793质谱仪、青岛海洋化工厂生产的柱色谱硅胶(300~400目)及高效薄层板。试剂均为市售分析纯或化学纯产品,除特别说明外,未经处理直接使用。2.2.15细胞为 HBV DNA克隆转染人肝癌细胞。
1.2 试验方法
1.2.1目标化合物3a、3b和4a~4c合成 参照文献[8]的方法,取L-酪氨酸甲酯盐酸盐1.1 mmol与 2-氯苯甲酸(4-氟苯甲酸或4-氯苯甲酸)1.0 mmol于反应瓶中,加入无水二氯甲烷(DCM)50 mL和N-甲基吗啉(NMM)2.3 mmol搅拌溶解完全,于0 ℃氩气保护下滴加含氯甲酸异丁酯(IBCF) 126 μL(1.0 mmol)和DCM 2 mL的混合溶液,0 ℃继续搅拌5 h。减压回收DCM,残留物用乙酸乙酯和水分散,萃取;所得有机层依次以蒸馏水、稀盐酸、饱和碳酸氢钠、饱和氯化钠洗涤,无水硫酸钠干燥,减压蒸干;将所得固体物溶解于N,N-二甲基甲酰胺(DMF)2 mL中,加入1.0 moL/L氢氧化钠溶液3 mL,室温搅拌1 h,反应液以浓盐酸调至pH 2~3,加入乙酸乙酯和水,萃取;所得有机层依次以蒸馏水、饱和氯化钠洗涤,无水硫酸钠干燥,减压浓缩至干即得中间体N-(2-氯-苯甲酰基)-L-酪氨酸(2a)、N-(4-氟-苯甲酰基)-L-酪氨酸(2b)、N-(4-氯-苯甲酰基)-L-酪氨酸(2c);将前一步合成的中间体2a1.0 mmol与L-苯丙氨酸甲酯盐酸盐1.1 mmol于反应瓶内,加入无水DCM 50 mL和NMM 2.3 mmol搅拌溶解完全,于0 ℃氩气保护下滴加含IBCF 126 μL(1.0 mmol)和DCM 2 mL的混合溶液,后0 ℃继续搅拌5 h,反应液减压回收DCM,剩余物以乙酸乙酯和水分散,萃取,所得有机层依次用蒸馏水、稀盐酸、饱和碳酸氢钠以及饱和氯化钠洗涤,无水硫酸钠干燥,减压蒸干,所得固体经硅胶柱层析纯化得状目标化合物3a。参照合成3a的方法可制备得3b和4a~4c。合成路线见图2。
1.2.1.1N-[N-(2-氯苯甲酰基)-L-酪氨酰基]-L-苯丙氨酸甲酯(3a) 白色粉末,收率74.5%。EI-MSm/z: 480 (M+),325,274,180,163,139 (100),120,107,91,77;1H-NMR (400 MHz,DMSO-d6)δ:9.20 (s,1H),8.52 (d,J=8.8 Hz,1H),8.43 (d,J=8.0 Hz,1H),7.46~7.16 (m,9H),7.07 (d,J=8.4 Hz,2H),6.66 (d,J=8.4 Hz,2H),4.64 (m,1H),4.53 (m,1H),3.59 (s,3H),3.07-2.67 (m,4H);13C-NMR (100 MHz,DMSO-d6)δ: 171.8,171.2,165.9,155.8,137.0,136.4,130.8,130.2,130.0,129.6,129.2,128.9,128.3,127.8,126.9,126.6,114.8,54.4,53.8,51.9,36.7,36.5。
1.2.1.2N-[N-(4-氟苯甲酰基)-L-酪氨酰基]-L-苯丙氨酸甲酯(3b) 白色粉末,收率68.4%。EI-MSm/z: 464 (M+),325,258,180,163,147,123 (100),95;1H-NMR (400 MHz,DMSO-d6)δ: 9.20 (s,1H),8.55 (m,2H),7.86 (m,2H),7.30~7.13 (m,7H),7.04 (d,J=8.4,2H),7.02 (d,J=8.4,2H),4.64 (m,1H),4.50 (m,1H),3.59 (s,3H),3.09~2.79 (m,4H);13C-NMR (100 MHz,DMSO-d6)δ: 171.8,171.8,165.1,162.7,155.7,137.1,130.6,130.5,130.2,130.1,130.0,129.1,128.3,126.6,115.2,115.0,114.8,54.9,53.7,51.9,36.6,36.2。
1.2.1.3N-[N-(2-氯苯甲酰基)-L-酪氨酰基]-L-苯丙氨醇(4a) 淡黄色粉末,收率84.9%。EI-MSm/z: 452 (M+),434,297,274,206,139 (100),120,107,91,77;1H-NMR (400 MHz,DMSO-d6)δ: 9.19 (s,1H),8.50 (d,J=8.4 Hz,1H),7.78 (d,J=8.8 Hz,1H),7.46~7.16 (m,9H),7.04 (d,J=8.4 Hz,2H),6.65 (d,J=8.4 Hz,2H),4.83 (t,J=5.6 Hz,1H),4.56 (m,1H),3.91 (m,1H),3.34~3.24 (m,2H),2.91~2.85 (m,2H),2.74-2.63 (m,2H);13C-NMR (100 MHz,DMSO-d6)δ: 170.5,165.8,155.7,139.0,136.5,130.8,130.2,130.0,129.6,129.2,128.9,128.1,127.9,126.9,125.9,114.5,62.1,54.8,52.3,36.8,36.4。
1.2.1.4N-[N-(4-氟苯甲酰基)-L-酪氨酰基]-L-苯丙氨醇(4b) 白色粉末,收率66.9%。1H-NMR (400 MHz,DMSO-d6)δ: 9.18 (s,1H),8.51 (d,J=8.4 Hz,1H),7.90~7.87 (m,3H),7.31~7.00 (m,6H),6.63 (d,J=8.4 Hz,2H),4.83 (t,J=5.6 Hz,1H),4.60 (m,1H),3.90 (m,1H),3.38~3.23 (m,2H),2.95~2.56 (m,4H);13C-NMR (100 MHz,DMSO-d6)δ: 171.3,171.1,165.0,162.7,155.7,139.1,130.1,130.0,129.2,128.4,128.1,125.9,115.2,115.0,114.8,69.8,55.2,52.5,36.5,36.4。
1.2.1.5N-[N-(4-氯苯甲酰基)-L-酪氨酰基]-L-苯丙氨醇(4c) 白色粉末,收率55.3%。EI-MSm/z: 452 (M+),434,297,274,206,139 (100),120,107,91,77;1H-NMR (400 MHz,DMSO-d6)δ: 9.15 (s,1H),8.54 (d,J=8.4 Hz,1H),7.87 (d,J=8.4 Hz,1H),7.81 (d,J=8.4 Hz,2H),7.53 (d,J=8.4 Hz,2H),7.25~7.12 (m,5H),7.07 (d,J=8.0 Hz,2H),6.60 (d,J=8.0 Hz,2H),4.80 (t,J=5.2 Hz,1H),4.57 (m,1H),3.88 (m,1H),3.35~3.24 (m,2H),2.95~2.60 (m,4H);13C-NMR (100 MHz,DMSO-d6)δ: 170.9,164.9,155.7,139.0,136.1,132.9,130.1,129.4,129.2,128.33,128.28,128.1,125.9,114.8,62.2,55.2,52.5,36.5,36.4。
1.2.2目标化合物5a~5e的合成通法 取化合物4c1.0 mmol与碳酸钾3.0 mmol于反应瓶内,加入DMF 10 mL,搅拌下加入碘甲烷1.1 mmol,室温搅拌至反应完全,将反应液分散于乙酸乙酯和水中,萃取,所得有机层依次以水、饱和氯化钠洗涤,无水硫酸钠干燥,减压蒸干,所得固体经硅胶柱层析纯化即得5a。同法,以碘代乙烷、碘代丙烷、碘代异丙烷或二甲氨基氯乙烷盐酸盐替换碘甲烷,可分别制得5b~5e。合成路线见图2。
1.2.2.1N-[N-(4-氯苯甲酰基)-O-甲基-L-酪氨酰基]-L-苯丙氨醇(5a) 白色粉末,收率76.5%。ESI-MSm/z: 955.5 [2M+Na]+;1H-NMR (600 MHz,DMSO-d6)δ: 8.57 (d,J=8.5 Hz,1H),7.90 (d,J=8.4 Hz,1H),7.84~7.78 (m,2H),7.55~7.50 (m,2H),7.25~7.15 (m,6H),7.16~7.09 (m,1H),6.82~6.76 (m,2H),4.83 (t,J=5.4 Hz,1H),4.64~4.57 (m,1H),3.92~3.84 (m,1H),3.67 (s,3H),3.33 (dd,J=10.6,5.1 Hz,1H),3.27 (dd,J=10.8,5.7 Hz,1H),2.95 (dd,J=13.8,4.5 Hz,1H),2.89~2.83 (m,2H),2.66 (dd,J=13.7,8.0 Hz,1H);13C-NMR (151 MHz,DMSO-d6)δ: 171.01,165.11,157.78,139.06,136.18,132.87,130.23,129.44,129.27,128.37,128.14,125.99,113.51,62.28,55.22,54.95,52.54,36.47。
1.2.2.2N-[N-(4-氯苯甲酰基)-O-乙基-L-酪氨酰基]-L-苯丙氨醇(5b) 参考5a合成方法,以碘乙烷替换碘甲烷合成目标化合物5b,白色粉末,收率60.6%。ESI-MSm/z: 503.2 [M+Na]+;1H-NMR (600 MHz,DMSO-d6)δ: 8.56 (d,J=8.5 Hz,1H),7.88 (d,J=8.4 Hz,1H),7.85~7.79 (m,2H),7.57~7.50 (m,2H),7.25~7.16 (m,6H),7.16~7.09 (m,1H),6.81~6.75 (m,2H),4.79 (t,J=5.5 Hz,1H),4.66~4.59 (m,1H),3.96~3.87 (m,3H),3.38~3.31 (m,1H),3.31~3.26 (m,1H),2.96 (dd,J=13.8,4.6 Hz,1H),2.88 (dd,J=8.0,2.4 Hz,1H),2.86 (dd,J=7.9,2.1 Hz,2H),2.67 (dd,J=13.6,8.0 Hz,1H),1.28 (t,J=6.9 Hz,3H);13C-NMR (151 MHz,DMSO-d6)δ: 171.38,165.48,157.46,139.48,136.55,133.32,130.62,130.51,129.84,129.66,128.75,128.53,126.36,114.38,63.24,62.68,55.60,52.94,36.88,15.15。
1.2.2.3N-[N-(4-氯苯甲酰基)-O-丙基-L-酪氨酰基]-L-苯丙氨醇(5c)制备 参考5a合成方法,以碘丙烷替换碘甲烷合成目标化合物5c,白色粉末,收率71.1%。ESI-MSm/z: 517.2 [M+Na]+;1H-NMR (600 MHz,DMSO-d6)δ: 8.56 (d,J=8.5 Hz,1H),7.89 (d,J=8.3 Hz,1H),7.83 (d,J=8.2 Hz,2H),7.53 (d,J=8.2 Hz,2H),7.26~7.16 (m,6H),7.13 (t,J=7.6 Hz,1H),6.79 (d,J=8.2 Hz,2H),4.79 (t,J=5.5 Hz,1H),4.62 (td,J=9.4,4.4 Hz,1H),3.91~3.86 (m,1H),3.84 (t,J=6.6 Hz,2H),3.37~3.32 (m,1H),3.31~3.26 (m,1H),2.96 (dd,J=13.9,4.5 Hz,1H),2.87 (dd,J=14.0,8.0 Hz,2H),2.67 (dd,J=13.8,8.0 Hz,1H),1.71~1.64 (m,2H),0.94 (t,J=7.4 Hz,3H);13C-NMR (151 MHz,DMSO-d6)δ: 171.38,165.50,156.35,139.48,136.55,133.33,130.66,130.40,129.84,129.66,128.74,128.52,126.36,115.62,69.36,62.68,55.60,52.95,36.91,36.88,22.31。
1.2.2.4N-[N-(4-氯苯甲酰基)-O-异丙基-L-酪氨酰基]-L-苯丙氨醇(5d)的制备 参考5a合成方法,以碘代异丙烷替换碘甲烷合成目标化合物5d,白色粉末,收率60.9%。ESI-MSm/z: 517.2 [M+Na]+;1H-NMR (600 MHz,DMSO-d6)δ: 8.57 (d,J=8.5 Hz,1H),7.88 (d,J=8.4 Hz,1H),7.85~7.81 (m,2H),7.56~7.50 (m,2H),7.25~7.17 (m,6H),7.16~7.10 (m,1H),6.80~6.74 (m,2H),4.79 (t,J=5.5 Hz,1H),4.66~4.59 (m,1H),4.56~4.48 (m,1H),3.96~3.86 (m,1H),3.38~3.32 (m,1H),3.31~3.26 (m,1H),2.96 (dd,J=13.9,4.7 Hz,1H),2.92~2.83 (m,2H),2.67 (dd,J=13.7,8.0 Hz,1H),1.21 (d,J=1.6 Hz,3H),1.20 (d,J=1.5 Hz,3H);13C-NMR (151 MHz,DMSO-d6)δ: 171.38,165.50,156.35,139.48,136.55,133.33,130.66,130.40,129.84,129.66,128.74,128.52,126.36,115.62,69.36,62.68,55.60,52.95,36.91,36.88,22.31。
1.2.2.5N-[N-(4-氯苯甲酰基)-O-二甲氨基乙基-L-酪氨酰基]-L-苯丙氨醇(5e) 白色结晶,收率46.6%。ESI-MSm/z: 525.2 [M+H]+;1H-NMR (600 MHz,DMSO-d6)δ: 8.57 (d,J=8.5 Hz,1H),7.89 (d,J=8.3 Hz,1H),7.85~7.80 (m,2H),7.57~7.51 (m,2H),7.26~7.16 (m,6H),7.16~7.11 (m,1H),6.84~6.77 (m,2H),4.80 (t,J=5.5 Hz,1H),4.66~4.59 (m,1H),3.96 (t,J=5.9 Hz,2H),3.94~3.87 (m,1H),3.38~3.31 (m,1H),3.31~3.27 (m,1H),2.97 (dd,J=13.8,4.5 Hz,1H),2.91~2.84 (m,2H),2.67 (dd,J=13.7,8.0 Hz,1H),2.56 (d,J=11.8 Hz,2H),2.18 (s,6H);13C-NMR (151 MHz,DMSO-d6)δ: 171.38,165.49,157.44,139.47,136.56,133.31,130.63,129.84,129.66,128.75,128.53,126.36,114.47,66.09,62.68,58.21,55.60,52.95,46.02,36.89。
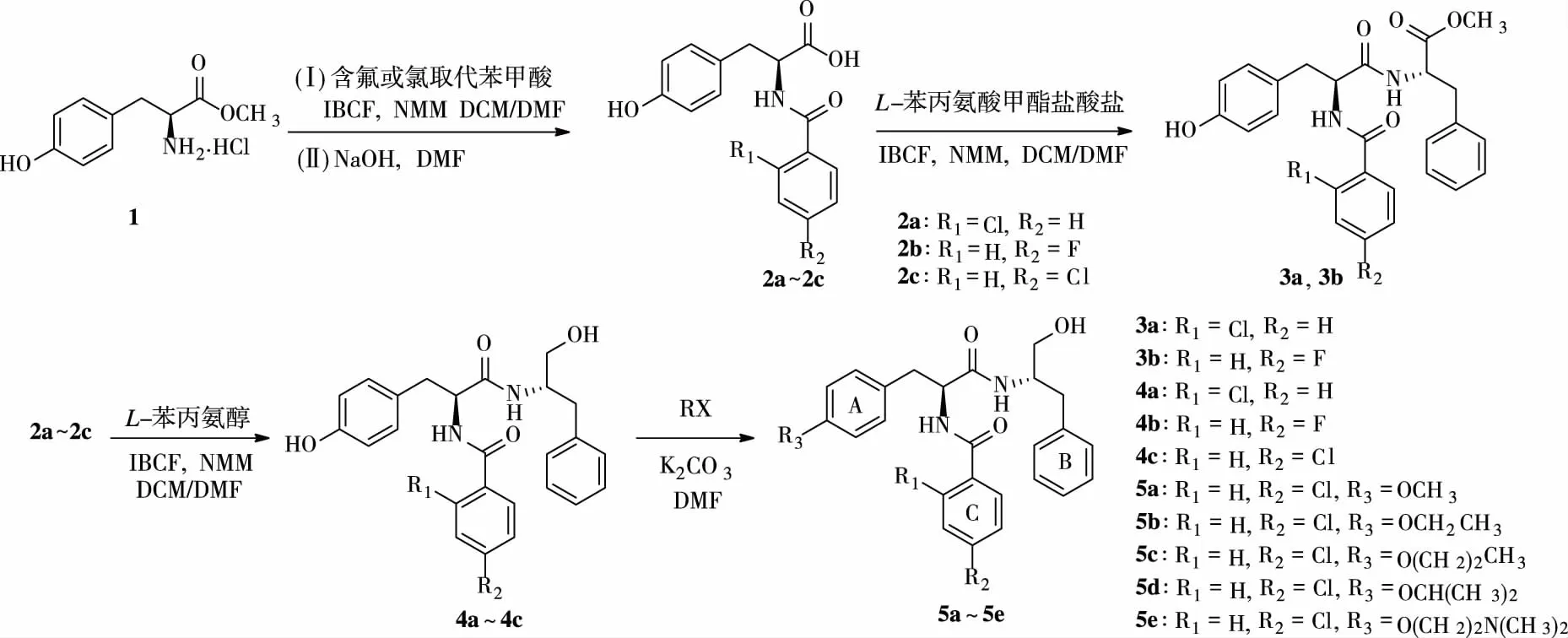
图2 目标化合物的合成路线图Fig.2 Synthetic route for target compounds
1.3 马蹄金素衍生物体外抗HBV活性研究
取2.2.15细胞接种96孔培养板,24 h后加入不同浓度的样品液(3a、3b、4a、4b、4c、5a、5b、5c、5d、5e、MTS)、空白对照液或拉米夫定(lamivudine)阳性对照液。培养3 d后,每孔加入5 g/L的MTT溶液20 μL,继续培养6 h,1 000 r/min离心10 min,吸去孔内培养液,每孔加入DMSO 200 μL,振荡10 min,形成的结晶充分溶解后、用酶联免疫检测仪测定其吸光度值(OD值),采用改良寇氏法计算药物的半数有毒浓度。另取2.2.15细胞接种96孔培养板,24 h后分别加入不同浓度的样品(3a、3b、4a、4b、4c、5a、5b、5c、5d、5e、MTS)及阳性对照药拉米夫定,同时设细胞对照孔,加药后每72 h分别更换含不同稀释浓度样品的培养液,于加药后第8日分别收集2.2.15细胞,采用斑点杂交的方法检测细胞中HBV DNA的复制程度,分别计算IC50及药物选择性指数(SI)[15]。
2 结果
通过化学方法合成了10个含氟或氯取代的MTS衍生物,并对所合成的目标产物进行体外抗HBV 活性测试,结果显示其中有8个目标产物对HBV DNA的复制有不同程度的抑制活性。化合物5a、5c、5d表现出良好的抗HBV活性,其IC50为12.61、10.53、6.46 μmol/L。见表1。
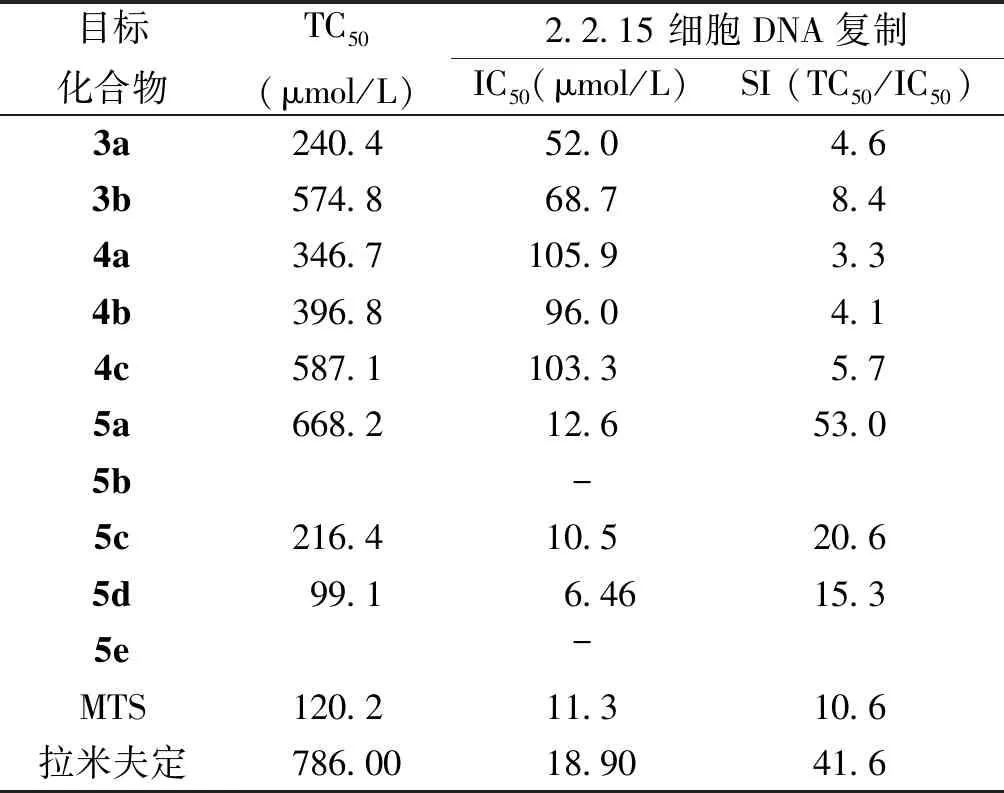
表1 目标化合物对2.2.15细胞DNA复制的抑制活性Tab.1 Inhibitory effect of the target compounds on HBV DNA in 2.2.15 cell
注:“-”表示样品在测试浓度范围内无活性
3 讨论
本文以具有抗HBV活性的MTS为先导化合物,通过在其B环引入氟或氯取代,设计合成了10个衍生物,由表1中目标化合物的抗HBV活性测试结果可以看出,A环为4-羟基取代的化合物活性相对较低;对化合物4c中A环4-位酚羟基进行甲基化、丙基化或异丙基化取代后所得化合物5a、5c、5d的抗HBV活性显着提高,特别是其中甲基化取代的化合物5a,其细胞毒性还略有降低,SI达到53.0,说明含氟或氯原子取代的MTS衍生物具有进一步研究的价值。



