邓皓锴 胥靓 伊木清
1上海体育学院(上海 200438)
2国家体育总局运动医学研究所(国家体育总局运动营养重点实验室,国家运动营养测试研究中心)
3北京体育大学
1 前言
内质网(endoplasmic reticulum,ER)是真核细胞内最大的亚细胞器,广泛分布于胞质并与多个亚细胞器联结,形成庞大的精密“膜网络系统”。ER在代谢激活、蛋白质合成与加工、Ca2+贮存和释放、胞质内物质运输等方面发挥作用,涉及的信号转导机制极为复杂,故ER功能失衡会累及细胞多种功能。当细胞内外环境发生变化时,细胞的应激反应涉及细胞核、线粒体、高尔基体等多种亚细胞器,但ER应激反应(endoplasmic reticulum stress,ERS)更为重要,且往往先于其他亚细胞器的应激反应。
多种病理生理状态如体温升高、局部组织和器官相对缺血缺氧、糖浓度降低、体液pH值降低、细胞Ca2+稳态失衡等因素均可诱发ERS。适度ERS有利于保护细胞,使ER在蛋白质过度合成与错误折叠、Ca2+贮存耗竭等状态下维持细胞内稳态和功能。而ERS持续存在或强度过大时,可通过三个ER跨膜感受器激活未折叠蛋白反应(unfolded protein response,UPR)信号通路,引起氧化应激、炎症和凋亡反应,这会加重或促发代谢性疾病(肥胖、代谢综合征)、心血管疾病、癌症、神经退行性疾病、骨质疏松症和肾脏疾病等病症[1-6]。
运动与ERS以及ERS与疾病和健康的关系是运动与健康及生物医学领域的研究热点,大量研究表明:运动能通过多种机制有效改善各种ERS应激相关的病症,包括肥胖、糖尿病、神经退行性病变、肌肉衰减综合征等[6]。
2 ERS的发生机理
2.1 ERS激活通路
ERS可通过三条信号通路激活:错误折叠蛋白或未折叠蛋白在ER集聚过多无法清除而引发的未折叠蛋白反应(UPR);细胞核因子κB(NF-κB)因折叠的蛋白质在ER中过多被激活而诱导的ER过载反应(endoplasmic reticulum-overload response,EOR);由胆固醇缺乏所引起的固醇调节级联反应。
2.1.1 UPR
UPR启动早期可起到保护细胞的作用,但持续时间过长或过强致ER无法应对时,将启动细胞程序性死亡即细胞凋亡。
研究表明,ER膜上蛋白肌醇必需酶1(inositol-requiring enzyme 1,IRE1)、蛋白激酶R型ER激酶(protein kinase R-like ER kinase,PERK)、活化转录因子(activating transcription factor 6,ATF6)在转录和翻译水平介导UPR的3条信号通路(图1),这3个跨膜蛋白的激活与ER伴侣蛋白GRP78(glucose regulating protein 78)解离程度有关。
1)ATF6通路:ATF6是一个ER Ⅱ型跨膜蛋白,N端位于细胞质,C端位于ER内,相对分子质量为90000。该通路的独特性在于ATF6与 GRP78分离后通过囊泡的形式移位到高尔基体并被S1P(site-1 protease)和 S2P(site-2 protease)切割后激活并发挥其活性,诱导XBP1 mRNA的表达,从而上调多种ERS相关基因表达[7,8]。
2)PERK通路:PERK蛋白N末端位于ER腔,C末端位于细胞质,是ERⅠ型跨膜蛋白。与IRE1相同,当PERK结构域与GRP78解离后形成同源二聚体且自身磷酸化后激活 eIF2α(eukaryotic translation initiation factor2α)磷酸化,eIF2α磷酸化后蛋白翻译折叠,起始复合物中 GDP与 GTP的能量交换得到抑制,从而暂缓蛋白翻译折叠及运输,缓解了ER的折叠蛋白压力,避免了ER过载运转[7,9]。
3)IRE1通路:IRE1在胞内段具有RNA酶活性和激酶活性,是ERⅠ型跨膜糖蛋白。GRP78与IRE1结构域在ERS时解离,IRE1通过寡聚化及自身磷酸化反应形成同源二聚体并激活了IRE1 RNA酶活性,两者底物为 X-box结合蛋白1(X-box binding protein1,XBP1)mRNA,其作用主要是在ER合成正确蛋白质及协调稳定折叠过程。IRE1 RNA酶活性切割XBP1 mRNA,其转录翻译生成活性转录因子XBP1s(spliced XBP1),XBP1s继而移位胞核后上调ERS相关基因表达,同时编码后的XBP1蛋白上调了GRP78的转录活性[7]。
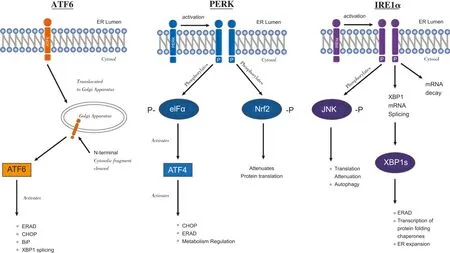
图1 UPR的跨膜调节因子[7]
2.1.2 EOR
ER过载反应的作用是激活 NF-κB转录因子,激活的机制可能与ERS时活性氧(ROS)生成及ER摄取和释放Ca2+有关,它是一条相对独立的ERS信号通路。
2.1.3 固醇调节级联反应
ERS发生时ER膜上的固醇调节元件结合蛋白(sterol regulatory element binding protein,SREBP)因ER膜上胆固醇的消耗被激活,随后SREBP与膜上SREBP裂解激活蛋白(SREBP cleavage-activating protein,SCAP)形成复合物水解为转录因子进入细胞核,从而增强靶基因的转录翻译[10,11]。
2.2 ERS标志蛋白
细胞应激蛋白(stress response protein,SRP),又称作“分子伴侣(molecular chaperones)”蛋白,是具有高度保守性细胞结构的机体应激非特异性产物。
ERS中最主要的分子伴侣蛋白是GRP78,是ER中的稳态感受器,对ER稳态的维持起着重要作用[12,13]。ER处于稳态时GRP78分别与IRE1、PERK、ATF6结合使这三种蛋白处于失活状态。ERS时GRP78迅速识别到未折叠蛋白或错误折叠蛋白并与这些蛋白结合,与此同时,GRP78与IRE1、PERK、ATF6蛋白解离激活了它们的活性,从而启动细胞应激机制。
3 ERS与氧化应激和细胞凋亡
3.1 ERS与氧化应激
氧化应激是指机体氧化与抗氧化能力的动态失衡,这是导致疾病与衰老的一个重要因素。ER为蛋白质提供了一个独特的氧化环境以促进二硫键的形成[14],蛋白质二硫键异构酶(PDI)可催化蛋白质的氧化折叠同时自身被还原,并由ER氧化还原蛋白1(ERO1)使其重新氧化[15],随着二硫键形成,电子从还原的PDI转移到分子氧(O2)形成H2O2,多个二硫键的氧化会产生高水平的ROS,仅由ER产生ROS的量就占ROS总量的25%[16]。也有证据表明,当ER中未折叠蛋白的积累通过IP3R引起Ca2+渗漏进入胞质时,也可能产生ROS[17]。在ER腔中蛋白质折叠是高能量消耗的过程,因此,细胞内ATP的消耗刺激线粒体氧化磷酸化导致ATP和ROS的产生[18]。
3.2 ERS与细胞凋亡
细胞凋亡是细胞的重要生理功能,凋亡在不同种属间高度保守。研究表明,细胞凋亡功能丧失可促进自身免疫系统疾病和肿瘤的发生,但如果细胞凋亡功能过强可引发退行性改变,如神经系统退行性疾病和免疫缺陷等。ERS持续时间过长或强度过大时,ER会通过UPR进一步强化IRE1、PERK、ATF6三条通路的应激反应,启动细胞凋亡程序。UPR的跨膜分子调节机制已于“2.1.1 UPR”中叙述,凋亡的信号通路如下。
3.2.1 CHOP通路
ERS状态下PERK通路和ATF6通路通过诱导表达CHOP/GADD153(C/EBP-homologousprotein/growth arrest and DNA damage-inducible gene 153)转录因子来抑制GRP78的表达,同时激活的ERO1可编码一个ER氧化酶,使ER处于富氧环境。CHOP还参与调节下游凋亡相关基因表达并引发细胞周期停滞和DNA的损伤,最终导致细胞凋亡。此外,抗凋亡蛋白Bcl-2因CHOP与cAMP反应元件结合蛋白(CREB)结合形成二聚体而被抑制表达[19]。
3.2.2 IRE1通路
ERS持续存在或过强时IRE1募集肿瘤坏死因子受体相关因子2(TRAF2),TRAF2与c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)和应激激活蛋白激酶(SAPK)偶联从而激活凋亡信号调节激酶1(ASK1)并且JNK也可以直接被IRE1激活后自身磷酸化为p-JNK,进而启动细胞凋亡(CHOP和JNK均是激活线粒体凋亡通路的上游信号)。同时,TRAF2与仅产生于ER的半胱天冬酶12(Caspase-12)作用,进而继续激活其下游凋亡因子Caspase-9和Caspase-3,诱导细胞凋亡[20,21]。
值得一提的是,ER存在着多种蛋白质四级结构折叠相关的酶系统,包括糖基化酶、蛋白质二硫键异构酶等,研究表明这些蛋白功能与Ca2+的浓度变化密切相关,Ca2+可作用于ERS的多个环节并参与细胞凋亡[22,23]。
4 运动与ERS
4.1 运动对ERS的双向作用
Luz等[24]通过对高脂饲养大鼠进行8周游泳训练后,发现有氧运动可以明显减轻大鼠肝脏组织和睾丸脂肪中ERS状态,明显降低了促炎因子的表达,并且有效改善了胰岛素的敏感性。Bourdier和Bozi等[25,26]研究表明,平板运动训练可降低GRP78、p-PERK、p-eIF2α、ATF6、XBP1、CHOP、Caspase-3的表达从而改善大鼠心血管功能,减少心肌梗死。间歇性爬梯训练和热量摄入减少的组合可以降低ERS相关蛋白p-PERK和CHOP的表达水平,这些蛋白与心肌损伤具有显着相关性[27]。4周跑台运动也可改善高脂饮食诱导的肥胖小鼠的内皮功能障碍(血管阻力)[28]。3个月有氧运动使肥胖成人的皮下脂肪组织和外周血单个核细胞(PBMCs)中GRP78、p-IRE1和p-eIF2α的mRNA和蛋白水平降低[29]。
Kim等[30]研究发现3周高强度跑台运动可以加强高脂饮食肥胖小鼠脑组织中的ERS程度,下丘脑ERS程度最大,但并未诱导细胞凋亡。与低强度组和对照组相比,高强度训练5周可降低大鼠骨骼肌ERS及凋亡信号蛋白CHOP和ATF4表达[31]。低强度和高强度间歇运动均可显着改善II型糖尿病青少年血液GRP78水平[32]。小鼠下坡跑训练8周后,趾长伸肌和比目鱼肌中ERS应激蛋白p-IRE1、p-PERK和p-eIF2α的表达高于上坡跑,表明离心收缩引起的ERS比向心收缩严重、损伤程度也更重;但对于这两种骨骼肌类型,下坡跑后的恢复期这些ERS蛋白未完全正常化;在第8周训练结束时,上坡跑的小鼠仅上调比目鱼肌的GRP78、pPERK和p-eIF2α水平,表明下坡跑诱导的骨骼肌ERS可能还与趾长伸肌和比目鱼肌的病理状况有关[33]。Wu等[34]发现,ATF6α敲除小鼠急性运动后的有效恢复受损,而通过敲除CHOP从而阻断ERS相关途径的细胞凋亡可改善PGC-1α敲除小鼠的运动不耐受。胥靓等[35]研究耐力运动训练也只轻度增加了骨骼肌GRP78表达。在神经系统方面,Li等[36]研究表明,高脂饮食肥胖可以诱导SD大鼠的前额叶ERS,过量的ERS可能在降低神经可塑性相关蛋白的水平中起关键作用;有氧运动可明显逆转SD肥胖大鼠前额叶皮质中GRP78,p-PERK,peIF2α,Caspase-12,CHOP和Bax/Bcl-2的表达。最近有报道:有氧运动通过减少大鼠心肌梗死引起的UPR标志性蛋白、未折叠蛋白聚集以及多聚泛素化蛋白的水平重建大鼠心肌的蛋白质量控制,从而改善大鼠的运动能力和心脏功能[26]。而短期的有氧运动不足以诱导心肌ERS相关蛋白GRP78、CHOP的表达增加[37]。
可见,不同运动方式、强度和时间引起的不同的器官、组织与类型的ERS程度、持续时间、对机体的影响和转归目前尚无一致性结果。而一致的看法是:适度ERS诱导表达GRP78等分子伴侣有利于细胞的正常生理机能、使细胞恢复到应激前的稳态,而当ERS时间过长或程度过强时,则会启动细胞凋亡。
4.2 细胞Ca2+失衡诱发ERS
ER是细胞内最大的Ca2+存储库,参与骨骼肌细胞兴奋-收缩偶联;同时,胞内游离Ca2+参与了细胞内绝大多数信号转导和能量代谢刺激作用。运动至疲劳(或力竭)时,肌细胞内Ca2+浓度、ER储存和释放Ca2+的功能失衡[38],此为引起ERS的诱因之一。
胞质中大量Ca2+可致线粒体钙超载,线粒体膜通透性改变、细胞色素C释放,进而激活下游凋亡因子Caspase-9和Caspase-3[39]。研究观察到,引起细胞凋亡的因素恰恰与诱导ERS发生的因素相吻合,而其中Ca2+平衡紊乱可直接诱发ERS,引发UPR、EOR,激活凋亡通路。其中,EOR通过激活转录因子NF-κB来调控对多种炎性蛋白的转录表达。Caspase-12作为ER膜上特有的结构蛋白,ERS时细胞质Ca2+紊乱直接使Caspase-12前体裂解活化,激活了下游凋亡因子Caspase-9和Caspase-3,从而引发细胞凋亡[40]。同时,细胞色素C转移到ER并与IP3R作用形成正反馈,促进了细胞凋亡、引起肌肉蛋白质降解。
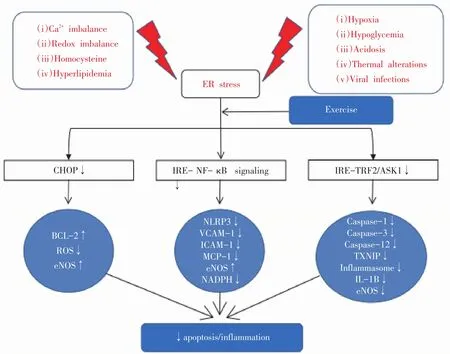
图2 运动训练改变ERS介导的凋亡和炎症反应(根据文献[4],有删节)
5 ERS与疾病和健康
5.1 ERS与肥胖
世界卫生组织估计超过19亿成年人体重超重,6亿多人肥胖,但有效和安全的药物治疗仍未成功[41]。肥胖的一个主要特征是瘦素抵抗,也可诱导下丘脑ERS[2],研究表明下丘脑ERS与瘦素抵抗之间存在因果关系。用ERS诱导剂(衣霉素、二硫苏糖醇等)处理的细胞和下丘脑切片显示:随着ERS增强其显着抑制瘦素诱导的信号传导及转录激活因子STAT3(signal transducers and activators of transcription)磷酸化[42,43]。此外,经侧脑室应用这些ERS诱导剂后所致的小鼠下丘脑ERS和瘦素抵抗与食物摄入量增加及体重增加呈正相关性[44-46]。
Shan[47]最近研究发现了一种ER功能障碍与代谢性炎症联系起来的新机制,该研究表明ERS通过IRE1α路径重新编程诱导脂肪组织巨噬细胞极化,导致全身葡萄糖体内平衡受损,加剧炎症和肥胖症状。Chen等[48]也发现高脂肪饮食诱导的肥胖有助于加强ERS和脂肪组织慢性炎症;同时随着ERS减轻,脂肪组织中游离胆固醇也随之减少。此外,4-PBA(4-phenyl butyric acid)和 TUDCA(ursodeoxycholic acid and its taurine-conjugated derivative)抑制肥胖小鼠体内脂肪组织中的NF-κB活性,降低炎症因子的表达,改善代谢紊乱。体外研究表明,IKK(IκB kinase)/NF-κB可能参与ERS诱导的脂肪因子分泌功能障碍的信号转导[2]。可以推测:抑制ERS可能是与肥胖相关代谢异常新药物靶标的新方向。
ERS可诱导胰岛素抵抗和糖尿病,最近,Ghemrawi等[49]对此进行了详细论述。肥胖、2型糖尿病和非酒精性脂肪肝病以及心血管疾病等虽有不同的生理和临床症状,但似乎具有营养过剩所致细胞和器官“糖毒性”和“脂毒性”诱导的共同病理特征---细胞内应激反应和炎症,包括ERS。
5.2 ERS与心血管疾病
多项研究表明ERS在心血管疾病的发病过程中起着重要作用[50-52]。内皮细胞(endothelial cell,EC)是维持血管稳态和控制内皮源性舒张因子(endothelium-derived relaxing factors,EDRFs)对血管敏感性的核心[53]。
在动脉粥样硬化中,UPR不能控制ER错误折叠蛋白量,并随着主动脉粥样硬化发展,CHOP的表达增加,最终激活CHOP诱导的细胞凋亡信号。PERK和ATF6通路通过调节CHOP来介导促凋亡bZIP转录因子表达,并通过多种途径激活IRE1通路的下游凋亡信号[54]。IRE1与TRAF2可以相互作用,其复合物与ASK1相联系,从而激活了JNK和p38丝裂原活化蛋白激酶(mitogenactivated protein kinases,MAPK)[52,55]。Bcl-2家族基因是控制CHOP和IRE1激活后凋亡和抗凋亡信号平衡的重要因子[56],也是ER和线粒体之间复杂的信号转导因子。由于CHOP介导的促凋亡蛋白致使ECs中线粒体功能下降[57]以及胞质中Ca2+的紊乱[3],使得动脉粥样硬化患者ECs ROS和烟酰胺腺嘌呤二核苷酸磷酸(NADPH)水平升高[58,59],但目前尚不清楚NADPH氧化酶是作为上游还是下游因子来调控ERS诱导的心血管功能障碍。
ERS时随着胞质Ca2+增多,ER膜上Caspase-12前体脱离膜结构转化为Caspase-12,这个过程需要钙蛋白酶参与激活下游的凋亡蛋白Caspase-9、Caspase-3[60]。缺血再灌注(IR)实验显示,心肌组织中Caspase-12和ER应激标志物水平升高[61-63];同时,在啮齿动物病理性心肌肥大模型中Caspase-12和Caspase-3的表达异常[64,65]。
ERS和炎症信号通路通过多种机制联系在一起也会引发心血管疾病。在动脉粥样硬化中,PERK、IRE1/TRAF2和ROS的增加激活并加重了炎症反应[66],二者通过募集IKK磷酸化IκB来调控炎症反应[67]。有研究指出:ATF6也与NF-κB-IKK相互作用,这表明ER三个应激感受器(PERK,IRE1和ATF6)可以通过ERS在细胞水平上增加炎症分子的表达如IL-8、IL-6等来诱导特异性炎症反应,从而引发动脉粥样硬化[68,69]。
最近Prola[70]等报道:ERS引起心肌细胞线粒体Ca2+摄取受损、线粒体氧化磷酸化功能改变以及从脂肪酸到糖底物消耗转移为特征的代谢重塑,故认为大多数心脏疾患的能量代谢受损应归因于ERS。
5.3 ERS与癌症
癌细胞因为其高增殖率需要高蛋白折叠能力的ER伴侣蛋白;同时,肿瘤微环境如缺氧、氧化还原失衡、pH波动和营养缺乏等是UPR的诱发因素[71]。
研究报道,UPR信号蛋白在癌细胞中表达上调[72],ER伴侣蛋白GRP78在前列腺癌、肺癌、乳腺癌和结肠癌中都有高表达[73,74]。此外,不表达GRP78的细胞不能形成肿瘤[75]。以上研究证实了GRP78在肿瘤形成中的重要作用。从本质上讲,GRP78增加了ER的蛋白质折叠能力,从而减少癌细胞中的应激所诱发的细胞凋亡。而UPR中的PERK在肿瘤增殖和存活中也起着重要作用,PERK通路失活是通过在PERK激酶结构域中产生突变或在极端缺氧情况下引入抗磷酸化的eIF2α形式损害细胞存活[76]。同时,PERK通过激活Nrf2限制氧化性DNA损伤,促进癌细胞增殖和存活[77]。PERKATF4介导的自噬也为癌细胞增殖提供氨基酸等营养物质[4,78]。此外,eIF2α的磷酸化为细胞提供了广泛的保护作用,因此其去磷酸化可能有助于肿瘤细胞凋亡。Dalton发现[79]GADD34的低表达与在恶性间皮瘤的侵袭性增加有关。单宁酸通过激活ERS调节相关蛋白ATF4、GRP78和PDI的表达,上调凋亡蛋白Bax、Bim和Caspase-3表达的同时下调了抗凋亡蛋白Bcl-2和BclxL的表达,抑制前列腺癌细胞的生长增殖[80]。以上研究提示:抑制ERS可能作为增强癌症治疗效果的新靶向。另外,XBP1s和GRP78在pADC(pulmonary adenocarcinoma,肺腺癌)中表达不同,但其过表达与患者预后不良有关,是pADC中独立的不良预后因素[81]。因此,ERS途径可作为生物标志物来判断癌症患者的预后。
5.4 ERS与骨质疏松症
骨质疏松症是老年人的主要健康问题,其特点是骨强度降低,易骨折。骨质疏松症的低骨密度(BMD)与ERS有关。PERK-eIF2α信号通路是胰腺和骨骼系统生长发育、功能和生存能力所必需的[5,82]。Liu 等[83]发现,在ERS状态下,eIF2α的磷酸化与交替单倍型相比,低BMD单倍型是显着增加的,这是由于相关的单核苷酸多态性变化引起的。He等[84]研究表明,通过阻断eIF2α去磷酸化可促进成骨细胞分化,他们还假设通过eIF2α和ATF4调节ERS可能是抗骨质疏松一个很好的治疗方式。Salubrinal作为eIF2α磷酸化抑制剂缓解ERS,减少破骨细胞的形成,抑制它们的迁移和粘附,并增加了GRP78、p-eIF2α和ATF4的表达,从而改善废用性骨质疏松症[85]。同时,IRE1α-XBP1通路是成骨细胞成熟的关键,在病理条件下的骨形成和骨吸收中起重要作用[86]。在成骨细胞中,Atg7缺乏引发ERS,通过施用苯基丁酸减弱ERS从而消除Atg7消融介导的对成骨细胞分化、矿化和骨形成的影响;同样,Atg7缺乏阻碍成骨细胞矿化并且通过CHOP和MAPK/JNK1-SMAD1/5/8依赖性方式促进部分细胞凋亡;而重建Atg7则改善ERS并恢复骨骼平衡[87]。此外,有报道称,从骨质疏松患者获得的成骨细胞中,ER特异性分子伴侣如GRP78和PDI表达量下调[88]。Liu等[89]研究表明大鼠成骨细胞中镉通过磷酸化钙调蛋白激酶(CaMKII)激活UPR并通过Caspase-12信号途径实现细胞凋亡。同样,抑制ERS和cAMP反应元件结合蛋白H(CREBH)信号通路阻断了破骨细胞的生成[90]。这些研究结果表明ERS在骨质疏松症、骨骼发育中的重要性,以及从ERS角度制定针对骨骼疾病的相关治疗策略的可能性。
5.5 ERS与神经退行性病变
神经退行性疾病的原因是多因素的,包括氧化还原失衡、环境因素、遗传易感性、谷氨酸诱导的兴奋毒性、神经功能紊乱、Ca2+稳态破坏、线粒体功能障碍和错误折叠蛋白质的积累等。ER应激蛋白IRE1和PERK被早老素蛋白(presenilin,PS)抑制,这是一个完整的膜蛋白和一部分分泌酶复合体,在ER和高尔基体中均有广泛表达[91]。研究表明,突变的PS降低了PERK-eIF2α通路的磷酸化,从而导致ER中蛋白质的积累[19]。错误折叠蛋白积累或未折叠蛋白引发的ERS有助于神经细胞凋亡[6,92]。IRE1α/XBP1作为UPR最保守的信号通路及UPR信号通路中关键调节因子,常在神经元细胞中被激活导致最终的神经退行性病变[93]。敲除XBP1基因能有效抑制通过立体定向注射6-OHDA诱发应激的多巴胺能神经元细胞的死亡。过表达人类α-突触核蛋白引起的ERS通过激活PERK和ATF6信号通路来上调凋亡蛋白CHOP在黑质多巴胺能神经元中表达,但可以通过过表达GRP78来抑制CHOP表达[94]。另外,ATF4持续激活并不抑制人类α-突触核蛋白表达引起的神经退行性疾病,但在帕金森病大鼠模型中能诱导多巴胺能神经元细胞大量凋亡[95]。Smith等[96]研究指出,α-突触核蛋白突变型 A53T异常表达使得ERS应激相关蛋白GRP78和CHOP表达升高,eIf2α磷酸化水平上调,但通过抑制 eIf2α磷酸化抑制ERS来保护α-突触核蛋白 A53T突变型过表达。近来,Coppola-Segovia等[97]提出了一种新的PD(parkinson's disease)模型,即通过腹腔注射衣霉素,在啮齿动物模型中观察到由于ERS作用导致的运动障碍、α-突触核蛋白寡聚化和多巴胺能神经元丢失等特征,这说明RES增强可能是PD发生发展的重要因素。
6 总结与展望
ER对细胞内外环境的微变化具有非常敏锐的洞察力,细胞稳态失衡时ERS立即启动,通过激活UPR信号通路调控内部环境,抵御应激,从而维持细胞的生存。但当刺激过大或ERS持续时间过长时,则会启动ER途径下的细胞程序性死亡(细胞凋亡),这一系列的生理性变化也是各种疾病发生的物质基础,ERS的程度直接关系到多种病症的发生发展。适度运动可减轻机体各器官细胞的ERS程度、降低ERS相关蛋白表达,而高强度或者长时间、不恰当运动则会加强ERS程度甚至引起器官组织的病理改变。
运动改善健康的ERS之运动项目、器官组织及不同个体的生理与病理效应“阈值”值得探索。因此,ERS与运动、疾病及健康的关系和机理,仍然是生物医学与运动科学领域的研究热点。



