程开生,杨 祥,臧洪梅
(1.杭州锐思医药科技有限公司,浙江 杭州 310052;2.安徽医科大学药学院,安徽 合肥 230032)
◇药物分析◇
GC法检测布洛芬注射液中杂质F
程开生1,杨 祥1,臧洪梅2
(1.杭州锐思医药科技有限公司,浙江 杭州 310052;2.安徽医科大学药学院,安徽 合肥 230032)
目的 建立气相色谱法测定布洛芬注射液中的杂质 F。方法 采用气相色谱法,色谱柱:DB-WAXetr毛细管柱(25 m× 0.53 mm ×2 μm),载气为氦气,流速5.0 mL·min-1,检测器为火焰离子化检测器,检测器温度为250℃,进样温度为200℃。结果 在选定的条件下布洛芬和杂质完全分离,杂质 F的检测限为 2.5 mg·L-1,检测方法的精密度、准确度、灵敏度均达到了预定的分析要求。结论 建立的方法准确、灵敏,适用于检测布洛芬注射液中的杂质 F。
气相色谱法;布洛芬;杂质
布洛芬是一种临床上广泛使用的非甾体抗炎药,属于芳基烷酸类药物,主要用于感冒、风湿和类风湿关节炎等疾病中炎症和疼痛的控制。该药剂型种类多,但国内目前均为布洛芬片剂、混悬剂、胶囊剂等口服制剂[1-2],为了解决难以口服药物患者的用药需求,研制布洛芬静脉注射液用于治疗疼痛和发热具有一定的临床意义。文献报道布洛芬中可能存在的有关物质有10余种,其中两种降解产物具有成纤维细胞和红细胞毒性[3-4]。国外药典采用 HPLC方法对布洛芬原料中各种有关物质设定了质量检查项,美国药典(USP34)[5]对杂质C、最大单杂和总杂进行了限定,英国药典(BP2009)[6]对杂质A、F、J、N作为特定杂质(specified impurity)控制,其中杂质F采用GC法检测,限度为0.1%。中国药典2010版[7]仅对布洛芬原料设置了自身对照的薄层色谱法检查有关物质,对布洛芬制剂均未设置有关物质检查项。注射液的质量标准要求比普通口服制剂要求高,特别是杂质限度,本文拟对制剂中杂质 F的限度和检测方法进行探讨,旨在为开发安全有效的布洛芬制剂提供借鉴。
布洛芬中的杂质 F,结构和布洛芬极为相似(见图1),采用薄层色谱和高效液相色谱不易将二者区分开来,英国药典 BP采用 GC方法对原料中的杂质F进行了含量测定,但是由于注射液含有水分,不利于GC的检测,因此笔者对该方法进行了改进,实验结果显示该方法快速、灵敏、准确度和重现性均较好,可以用于检测制剂中杂质F的含量,尤其是含水的制剂。
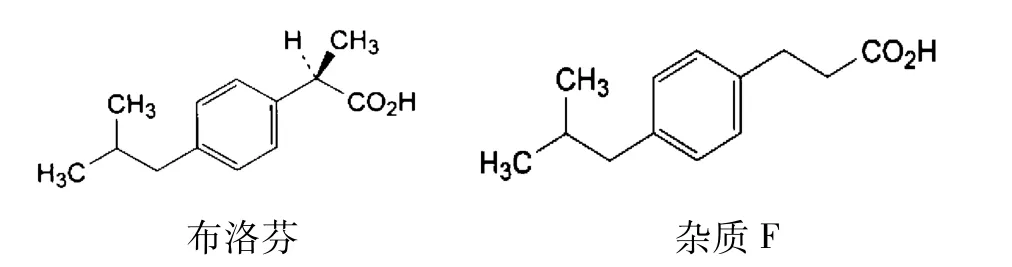
图1 布洛芬和杂质 F的结构
1 仪器与试药
安捷伦7890A气相色谱仪,包括安捷伦 chem-station色谱工作站、G7683B自动进样器(美国)等;Mettler-Toledo XS-105十万分之一电子天平。
乙酸乙酯(EtOAc)、N,N-二甲基甲酰胺二甲缩醛(DMF-DMA)、吡啶等为分析纯。布洛芬注射液自制(规格 8 mL:0.8 g,批号 110401、110402、110403;规格4 mL:0.4 g,批号 110404、110405、110406);布洛芬注射液市售(美国 Cumberland公司,4 mL:0.4 g,批号W029420AA)。布洛芬对照品(中国药品生物制品检验检定所,批号100179-200804,含量99.5%);杂质F对照品(EUROPEAN PHARMACOPOEIA REFERENCE STANDARD;LGC GmbH,批号:1.4;Cat.No.Y0000140)。
2 方法与结果
2.1 色谱条件 色谱柱:DB-WAXetr毛细管柱(25 m×0.53 mm×2 μm);载气:氦气,流速5.0 mL· min-1;检测器:火焰离子化检测器(尾吹:He:30 mL ·min-1,空气:300 mL·min-1,H2:30 mL·min-1);柱温:157℃;检测器温度:250℃;进样温度:200℃。
2.2 供试品溶液的配制
2.2.1 布洛芬注射液供试品溶液配制过程优化
(1)布洛芬提取方法优化:布洛芬注射液中的布洛芬与氨基酸成盐,采用盐酸将布洛芬置换沉淀出来,再用乙酸乙酯萃取沉淀即得布洛芬。
取本品适量(约相当于布洛芬800 mg)加稀盐酸适量酸化至无白色沉淀物出现,用乙酸乙酯 10 mL萃取,取乙酸乙酯层,氮气吹干,60℃干燥,称重,酸化萃取3次。见表1。
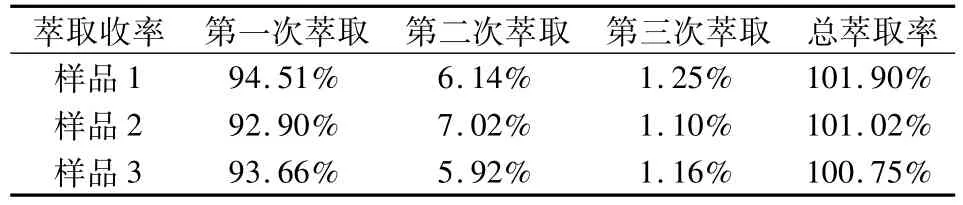
表1 酸化萃取优化结果
上述研究表明,加2 mL稀盐酸酸化后用乙酸乙酯萃取3次已基本萃取完全。
(2)乙酸乙酯萃取物纯度检查:按照布洛芬注射液注册标准中含量检查法(HPLC法)检查布洛芬注射液和乙酸乙酯萃取物中布洛芬的含量,结果含量均大于99%,符合要求。
(3)甲基化试剂用量的选择:取萃取次数选择项下干燥物约50 mg,加入甲基化溶液1.0、1.5、2.0 mL溶解,密封,在100℃加热20 min,冷却至室温,氮气吹干,将残留物用5 mL乙酸乙酯溶解,作为供试品溶液。分别取1 μL注入气相色谱仪,记录色谱图,考察不同用量甲基化试剂对衍生化反应的影响。结果峰面积分别为28 579、27 597、28 950,表明甲基化试剂1.0 mL能使反应顺利完成,加大用量不影响布洛芬峰面积。因此,甲基化试剂用量应为1.0 mL。
(4)衍生化时间选择:取萃取次数选择项下干燥物约50 mg,3份,分别加入1 mL的甲基化溶液溶解,密封,分别在100℃加热10、20、30 min,冷却至室温,氮气吹干,将残留物用5 mL乙酸乙酯溶解,作为供试品溶液。分别取 1 μL注入气相色谱仪,记录色谱图,考察不同反应时间对衍生化反应的影响。研究表明,反应20 min能使反应基本完成。
2.2.2 供试样品按配制方法 Ⅰ、DMF-DMA的乙酸乙酯溶液:取甲基化溶液1 mL,加入4 mL乙酸乙酯摇匀,即得。
Ⅱ、布洛芬乙酸乙酯溶液:取布洛芬对照品50 mg用5 mL乙酸乙酯溶解,即得。
Ⅲ、杂质F溶液:另取杂质F(内标液)1 mL加入1 mL的甲基化溶液溶解,密封,分别在100℃加热20 min,冷却至室温,氮气吹干,将残留物用5 mL乙酸乙酯溶解。
Ⅳ、系统适用性溶液:取布洛芬注射液(约相当于布洛芬800 mg),加杂质F内标液,再加稀盐酸2 mL酸化后,用乙酸乙酯萃取3次,每次10 mL,各份分别合并乙酸乙酯层,氮气吹干,60℃干燥。取干燥物约50 mg,加入1 mL的甲基化溶液溶解,密封,分别在100℃加热 20 min,冷却至室温,氮气吹干,将残留物用5 mL乙酸乙酯溶解,作为供试品溶液。
Ⅴ、精氨酸溶液:取精氨酸678 mg加水8 mL溶解,加稀盐酸2 mL酸化后,用乙酸乙酯萃取3次,每次10 mL,合并乙酸乙酯层,氮气吹干。结果无任何残渣,表明萃取过程中已排除了精氨酸的干扰。不再进样。2.3 专属性 按上述色谱条件,取上述溶液Ⅰ~Ⅳ,测定,结果见图2。Ⅰ在2~5 min有峰,与系统适应性对应的布洛芬峰和杂质F峰处无峰。Ⅱ在系统适应性对应的布洛芬峰处(tR=16.65 min)有峰,杂质F峰处无峰。Ⅲ在系统适应性对应的杂质F处(tR=25.55 min)有峰,布洛芬处无峰。Ⅳ在布洛芬和杂质 F分别出峰。结果表明,试剂、辅料均不干扰杂质F测定,本法专属性良好。
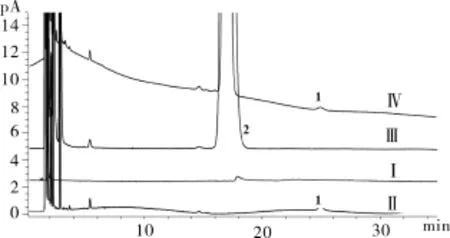
图2 气相色谱分离图
2.4 稳定性 系统适应性溶液在室温下放置,在0、18、24 h时,分别精密量取1 μL,注入色谱仪,记录色谱图。结果显示试验表明,杂质 F在18 h内稳定。
2.5 回收率 取本品9份,每份适量(约相当于布洛芬 800 mg),分别加杂质 F内标液 19.2 mL (120%浓度)、16 mL(100%浓度)、8 mL(50%浓度),分别再加稀盐酸 2 mL酸化后,用乙酸乙酯萃取3次,每次10 mL,各份分别合并乙酸乙酯层,氮气吹干,60℃干燥。取干燥物约 50 mg,加入 1 mL的甲基化溶液溶解,密封,分别在 100℃加热 20 min,冷却至室温,氮气吹干,将残留物用5 mL乙酸乙酯溶解,作为供试品溶液。每份均按上述方法配制。
另取杂质F(内标液)1 mL加入1 mL的甲基化溶液溶解,密封,分别在100℃加热20 min,冷却至室温,氮气吹干,将残留物用 5 mL乙酸乙酯溶解,作为杂质 F溶液。
分别取1 μL注入气相色谱仪,记录色谱图,考察回收率,结果见表2。
表 2 加样回收试验结果(n=3)
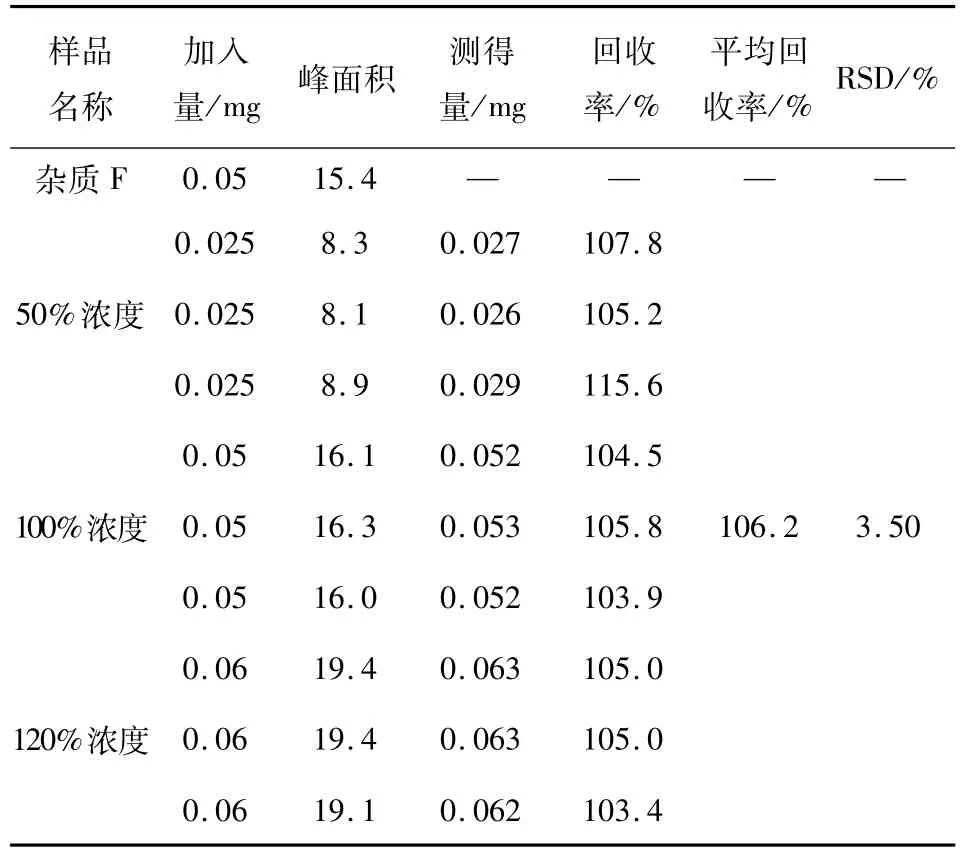
样品名称 量/mg 峰面积 测得量/mg加入 回收率/%平均回收率/%RSD/%杂质F 0.05 15.4— —50%浓度100%浓度106.2 3.50 120%浓度0.025 0.025 0.025 0.05 0.05 0.05 0.06 0.06 8.3 8.1 8.9 16.1 16.3 16.0 19.4 19.4 0.027 0.026 0.029 0.052 0.053 0.052 0.063 0.063 107.8 105.2 115.6 104.5 105.8 103.9 105.0 105.0 0.0619.10.062103.4
可见本法中,杂质F的平均回收率为 106.2%,回收率良好。
2.6 检测限 取回收率项下50%浓度样品1 mL,加入乙酸乙酯1 mL,摇匀,精密量取1 μL进样。结果信噪比约为3∶1,此法杂质F检测限为 2.5 mg· L-1。
2.7 杂质 F检查 取本品自制两个规格(六批)和市售两个规格(两批),按上述方法检验结果见表3。
研究表明,本品自制样品和市售样品的杂质 F均小于0.1%。
表3 布洛芬注射液有关物质测定结果(%)
3 讨论
英国药典(BP2009)中布洛芬的杂质F的GC检测方法适用于无水样品,因此笔者以BP2009为基础,优化布洛芬注射液中杂质 F的检查方法,并进行方法学验证。
布洛芬注射液为水溶液,不利于 GC毛细管柱分析;甲基化试剂(DMF-DMA)极易水解,布洛芬和精氨酸以盐的形式存在,不利于衍生化反应;因此需对供试样品配制方法进行改进:先用酸化法,将布洛芬和杂质 F将其置换出来,利用相似相溶原理,采用乙酸乙酯将布洛芬和杂质 F提取出来,得到其混合物,通过甲基(酯)化反应,利用布洛芬和杂质F甲基化产物沸点不同,通过 GC将其分离。
因此本文对供试品的配置中盐酸的用量和萃取的次数,提取物的纯度,衍生化条件进行考察。
文中所用的GC法测定精氨酸布洛芬注射液中杂质F的含量,其重复性好,操作简便,可用于订立精氨酸布洛芬注射液质量标准。杂质 F加样回收率大于100%可能与样品中含有低于检测限的杂质F有关。
[1] 裴小龙,宋愿智,杨 欣.布洛芬不同剂型微生物限度检查方法验证汇总[J].安徽医药,2013,17(10):1681-1682.
[2] 刘 瑞,刘福利,罗国平,等.HPLC法测定布洛芬注射液中精氨酸的含量[J].西北药学杂志,2013,28(4):361-365.
[3] Dondoni A,Dall'Occo T,Fanyin G,et al.Studies on the actual and potential impurities in ibuprofen[J].Farmaco Prat,1986,41(7):237-244.
[4] Castell JV,Gomez MJ,Miranda MA,et al.Photolytic degradation of ibuprofen:toxicity of the isolated photoproducts on fibroblasts and erythrocytes[J].Photochem Photobiol,1987,46(6):991-996.
[5] 美国药典委员会.美国药典(USP32 NF27)[S],2009:2607.
[6] 欧洲药品质量委员会.欧洲药典[S],6.8版,2008:5953.
[7] 国家药典委员会.中国药典:二部[S].北京:中国医药科技出版社,2010:120.
Determination of impurity F in ibuprofen injection by gas chromatography
CHENG Kai-sheng1,YANG Xiang1,ZANG Hong-mei2
(1.Hangzhou Raise Medical Technology Co.,Ltd,Hangzhou 310052,China;2.College of Pharmaceutics,Anhui Medical University,Hefei 230032,China)
Objective To establish a GC method to determine the impurity F in ibuprofen and its preparations.Methods The GC method was performed on a ZB-WAXetr capillary column(25 m×0.53 mm×2 μm)and FID detector.The carrier gas was He with flow rate at 5 ml/min.The injector temperature and the detector temperature were 200℃ and 250℃,respectively.Results Under the described chromatographic condition,The impurity F was completely separated from ibuprofen.The LOD of impurity F was 2.5 mg· L-1.Conclusions The analytical method is simple,accurate and sensitive with good reproducibility.It can be used to determine impurity F in ibuprofen and its preparations.
gas chromatography;ibuprofen;impurity
10.3969/j.issn.1009-6469.2014.05.009
2013-11-08,
2014-01-20)
科技部中小企业创新基金课题(No 09C26223304082);杭州市种子基金课题(No 20090531K10)



