, ,
(1.Key Laboratory of Basic Pharmacology and Joint International Research Laboratory of Ethnomedicine of Ministry of Education,Zunyi Medical University,Zunyi Guizhou 563099,China;2.Department of Drug Clinical Trial of The Third Affiliated Hospital of Zunyi Medical University, Zunyi Guizhou 563000,China)
[Abstract]One of the principal pathological features of the progression of Alzheimer’s disease(AD)is the occurrence of aberrant phosphorylation of the microtubule-associated protein tau(tau)in the AD brain.Moreover,it has been reported that abnormal hyperphosphorylation of tau protein is an early manifestation of AD and inhibition of tau protein phosphorylation is thought to be the key of AD therapy.However,the evolving mechanisms by which tau protein is hyperphosphorylated and accumulated are still not clear.Most importantly,the down-regulation of protein phosphatase 2A(PP2A)activity is thought to play an important role in the formation of tau protein hyperphosphorylation.Here,this review focuses on recent developments of tau protein hyperphosphorylation mainly involving in PP2A dysfunction.We also highlight that the evolving mechanism of PP2A dysfunction by which tau protein is hyperphosphorylated and contributes to its accumulation is reshaping the framework for the development of therapeutics targeting PP2A to treat AD.
[Key words]Alzheimer disease; protein phosphatase 2A; microtubule-associated protein tau; tau hyperphosphorylation
Alzheimer’s disease(AD)is characterized by two major pathological hallmarks,which include senile plaques(SPs)composed of extracellular deposits of amyloid β(Aβ)peptide and neurofibrillary tangles(NFTs)containing intracellular filamentous aggregates of hyperphosphorylated tau protein[1-2].Although a number of studies have focused on Aβ,clinical evidence has shown that anti-amyloid treatment for AD has failed to slow down the progression of the disease[3-4].In contrast to Aβ pathology,tau protein pathology is considered to correlate with the progression of the AD symptoms[5].Since then,attention has been shifted towards the research on the pathology of tau protein in AD.
Tau protein belongs to the family of microtubule-associated proteins,and mainly established function is to stabilize neuronal microtubules and promote their assembly,which is regulated by its degree of phosphorylation.However,a number of studies have shown that tau protein becomes hyperphosphorylated in AD.In addition,it has been reported that abnormal hyperphosphorylation of tau protein is an early manifestation of AD[6].Nevertheless,it is not completely understood the mechanisms contributed to tau protein hyperphosphorylation in AD.So far,there are many reported factors rendering tau protein phosphorylation.Among them,the disruption of this balance that tau protein phosphorylation is coordinated by the opposing actions of tau kinase and phosphatase activities is thought to be the main mechanism and to be at the origin of abnormal tau protein,thereby contributing to tau protein aggregates[7].Most importantly,protein phosphatase 2A(PP2A)is the most active phosphatase,accounting for about 70% of the human brain tau phosphatase activity[8].Accumulating evidence has shown that the activity of this enzyme is decreased in AD brain[9-11].Nevertheless,the mechanisms underlying the downregulation of PP2A activity in AD brain remain unclear.Here,we will more specifically discuss the role of the PP2A on tau protein pathology in AD.
1 Tau pathology
1.1 Tau structure and physiological function Tau protein,firstly separated by Weingarten from tubulin extracted from the porcine brain in 1975[12],is a microtubule-associated protein encoded by a single gene,microtubule-associated protein tau(MAPT)gene,which is located on chromosome 17 that contains 16 exons.Tau protein is found in adult cerebral cortex,hippocampus,and cerebellum,primarily located in the axons of nerve cells,whereas restricted to the somatodendritic compartment in AD[13],and most recently,a study showed that tau protein is highly expressed in pancreatic islets[14].In the adult human brain,as shown in Figure 1A,there are six tau protein isoforms which result from alternative mRNA splicing of exons 2,3,and 10[15].These isoforms differ from one another by the presence or absence of a 29-or 58-amino acid insert(encoded by exon 2 or 3,respectively)in the amino-terminal and the presence of three repeats(3R)or four repeats(4R)of 31 amino acids encoded by exon 10 in the microtubule-binding domain(MTBD)of the carboxy-terminal.Consequently,alternative splicing of tau mRNA yields the six tau protein isoforms called 0N3R,1N3R,2N3R,0N4R,1N4R and 2N4R,ranging from 352 to 441 amino acids in length[15-16].Besides,the central region of tau protein comprises the proline-rich domain(PRD)[17].Notably,the polypeptide chain of tau protein is not a compact but highly flexible,mobile and natively unfolded protein when binding to microtubule protein,which exhibits no secondary structure(α-helix,β-sheet,polyproline II helix),if any is only transient(Figure 1B)[18].In addition,when tau protein is free in the cytoplasm,it shows a preference for global interactions between domains that can be likened to a “paperclip” conformation within which the C-terminus folds over the MTBD and the N-terminus folds back over the C-terminus,bringing both termini in close proximity(Figure 1B)[16,19],likely preventing interactions with additional protein by masking the microtubule-binding repeats.Tau protein expression is developmentally regulated.In the normal adult human brain,all six isoforms of tau protein are expressed in the central nervous system(CNS).Moreover,there are similar levels of three-repeat and four-repeat tau protein isoform,and alteration of the 1/1 ratio of tau 3R/4R is sufficient to cause neurodegeneration and dementia[20].Tau protein isoforms containing three repeats are expressed throughout life including in the fetus,whereas only the shortest 0N3R-tau protein isoform,not but four repeats,is expressed in fetal brain[16,21-22].Goedert and Jakes[21]found the 4R tau protein isoforms promoted microtubule assembly at a rate 2.5-to 3.0-fold faster than the 3R tau protein isoforms,whereas the amino-terminal insertions did not appear to contribute to the assembly.
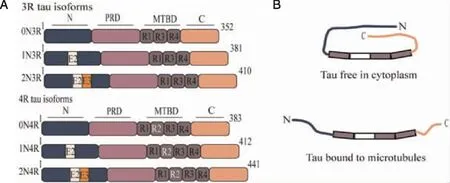
A:Tau protein domains and alternative splicing.Tau protein consists of four parts:the N-terminal region(N),the proline-rich domain(PRD),the microtubule-binding domain(MTBD)and the C-terminal region(C).Six tau protein isoforms are generated by alternative splicing of the MAPT gene,and these isoforms,ranging in size from 352-441 amino acids in human brain,differ by the absence or presence of one or two 29 amino acids inserts encoded by exon 2(E2,white box)and 3(E3,orange box)in the amino-terminal part and of the second microtubule-binding repeat(R2)encoded by exon 10 in MTBD.B:Tau protein structure in different status.The N-and C-termini of tau protein are closely associated when it is free in the cytoplasm,giving rise to the proposed “paperclip” conformation.On the other hand,when binding to microtubules primarily through the MTBD,the terminal regions of tau protein become separated.
In the CNS,tau protein has been shown to play an important role in the assembly of tubulin monomers into microtubules to constitute the neuronal microtubules network that serves as tracks for axonal transport under the physiological condition.For example,tau protein microinjected into fibroblast cells increased tubulin polymerization and stabilized microtubules[23].Additionally,some studies on tau deletion mice that do not express the tau protein have shown that it plays a pivotal role in learning,memory and synaptic plasticity[24-26],and embryonic hippocampal cultures from tau deletion mice showed a significant delay in axonal growth and neuronal maturation,suggesting an essential role for tau protein in neuronal maturation.Besides,tau protein might play a relevant role in DNA protection,regulating the cell cycle and lipid signaling.On the other hand,in some recent studies,using tau deletion and overexpression mice,researchers demonstrated the direct effects of tau protein on the periphery in regulating pancreatic β cell function and glucose homeostasis[14,27].Interestingly,elevated levels of abnormally phosphorylated tau protein occurred in pancreas tissue of patients with type 2 diabetes(T2D)and in pancreatic islets from a transgenic mouse model of AD and T2D[28-29].
1.2 Conformational changes of tau protein As described above,the structure of tau protein is important for its normal functions.In AD patients,tau protein exists in a misfolded state,and its misfolding would affect the microtubule binding and assembly,that is,tau protein is neither bound to tubulin nor promotes the microtubule assembly,therefore resulting in the deterioration of the organization of microtubule[15].In addition,studies have revealed that tau protein becomes hyperphosphorylated in AD brains,and its hyperphosphorylation leads to its detachment from microtubules and missorting into the somatodendritic compartment,thus increasing the amount of free tau protein within the cytoplasm,which then leads to its self-association into soluble non-filamentous and insoluble filamentous aggregates.Among them,tau protein species have been shown in the formation of the non-filamentous aggregates,including truncated tau protein[30],tau dimers[31],trimers[32]and soluble phosphorylated high-molecular-weight oligomers of tau protein(HMW-tau).The oligomeric tau protein lengthens into filamentous aggregates; namely,it adapts a β-sheet structure and transforms into granular tau oligomers under atomic force microscopy(AFM),and as these granular tau oligomers fuse together,they form paired helical filaments(PHFs),which ultimately form NFTs that are abundant in the brain of AD patients[31-33].A recent study suggested that soluble tau protein species can undergo liquid-liquid phase separation to form droplet-like tau protein observed in neurons and become gel-like in minutes,involving initiating tau protein aggregation[34].Remarkably,a number of studies have provided evidence that tau protein hyperphosphorylation precedes the dissociation of the protein from microtubules and leads to the loss of biological functions,the gain of toxic activity,and the aggregation into filamentous aggregates[35-36].Normal tau protein contains 2-3 moles of phosphate per mole of the protein,the level of phosphorylation for its optimal activity[37-38].However,in the autopsied AD brain,all of the six tau protein isoforms are present in a hyperphosphorylated state,and the level of tau protein phosphorylation is at least three-fold greater than that in the normal brain[39-40].Additionally,studies have shown that the largest human tau protein isoform 2N4R potentially has 85 putative phosphorylation sites,including 45 serines,35 threonines and five tyrosines,which are potentially phosphorylated in AD[38].Most importantly,findings from phosphorylation-dependent monoclonal antibodies against tau protein and mass spectrometric analysis have indicated that the total number of reported phosphorylation sites are more than 40 sites on PHF-tau from AD brain,and most occur in the proline-rich region and C-terminal tail region flanking the MT-binding repeats[36].Nevertheless,it is not completely understood the mechanisms contributed to tau hyperphosphorylation in AD.
2 PP2A:a major phosphatase

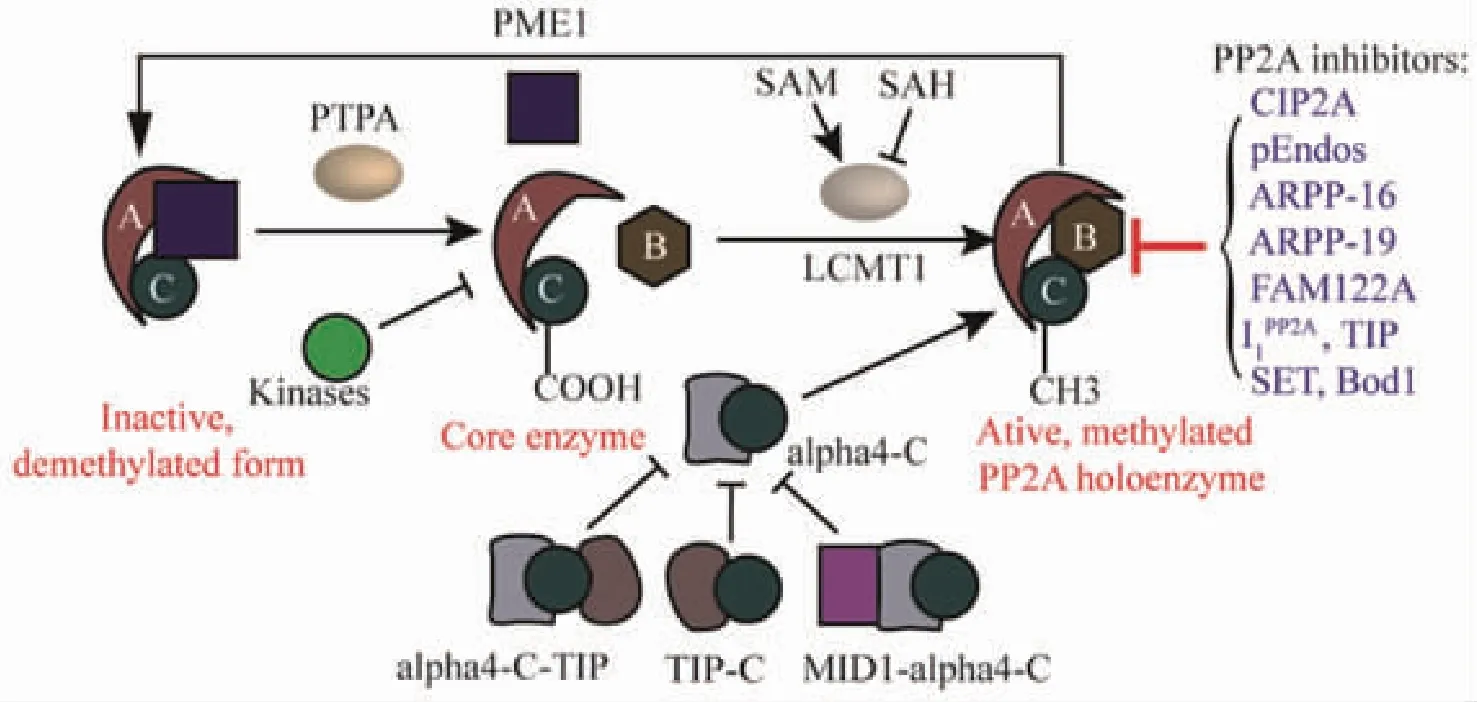
Fig 2 PP2A and its regulation
PP2A occurs in two distinct forms:heterodimeric form(core enzyme)composed of a PP2AC subunit and a structural A subunit and heterotrimeric form(holoenzyme)containing one additional regulatory B-type subunit.Several post-translational modifications of the highly conserved PP2A C tail constitute important determinants of holoenzyme assembly.In particular,reversible Leu309 methylation of PP2AC,which is catalyzed by the methyltransferase LCMT1,requires the supply of SAM and is inhibited by SAH,as well as demethylated and inactivated by the methylesterase PME-1.The inactive PP2A could be reactivated via the action of the PP2A activator PTPA,allowing for subsequent methylation of PP2AC.In addition,Thr304 and Tyr307 phosphorylation catalyzed by receptor and nonreceptor protein tyrosine kinases may also affect the recruitment of particular B-type subunits in the complex.In these cases,dephosphorylation is thought to occur by an autodephosphorylation mechanism.Besides,α4 binds to PP2AC to maintain active PP2A by preventing PP2AC ubiquitination,not relying on PP2AC methylation.MID1 protein normally targets PP2AC for degradation through binding to α4 thereby stimulating proteasomal degradation of PP2AC and inhibiting the activity of PP2A.TIP is an inhibitory regulator of PP2A by the forms of dimeric C-TIP and trimeric TIP-C-α4 complexes.Additionally,A number of endogenous PP2A inhibitors have been shown to either directly bind to the PP2AC or target very specific PP2A holoenzymes,thereby preventing dephosphorylation of a large variety of PP2A substrates(blue text).
2.2 Reversible PP2AC post-translational modifications The PP2AC subunit is the catalytically active component of the enzyme,and a highly conserved motif in the PP2A C tail,304TPDYFL309,plays an important role in regulating the assembly of PP2A complexes[51].Carboxyl methylation of the PP2AC is thought to play a critical role in the biogenesis of PP2A holoenzymes,uniquely methylated on leucine residue 309(Leu309)[52-53],and now is appearing to be a fundamental process for the regulation of many cellular processes[51],which is reversibly regulated by two specific enzymes with opposing activities,LCMT1 and PME-1[53-54](Figure 2).PP2AC is methylated and demethylated at the Leu309 by LCMT1 and PME-1,respectively.LCMT1 binds directly to the PP2AC active site and is responsible for the methylation of Leu309,whose activity depends on the supply of the universal methyl donor,S-adenosylmethionine(SAM),and is inhibited by S-adenosylhomocysteine(SAH)(Figure 2)[55].This methylation increases the binding affinity of core dimer towards distinct regulatory subunits,and this provides specific activities to the PP2A holoenzyme.Conversely,two inactive synthetic mutants of PP2A were shown to interact with PME-1,whereas active PP2A did not bind[56].The PP2A-specific methylesterase PME-1 can directly bind to the active site of the PP2AC,and remove the methyl group and inactivate PP2A by evicting manganese ions required for the phosphatase activity[57].However,past studies have shown that PME-1 is tightly associated with an inactive dimeric or trimeric form of PP2A,and these inactive enzyme forms could be reactivated as Ser/Thr phosphatase by PTPA through reducing its phosphorylation at Tyr307 with upregulation of protein tyrosine phosphatase 1B[58](Figure 2).Studies in cells have revealed that LCMT1 deficiency causes apoptotic cell death[59-60],and both LCMT1 and PME-1 knockout mice have lethal phenotypes[61-62],suggesting that these enzymes have indispensable functions.The methylated and demethylated forms of PP2A play a critical role in the maintenance of the enzyme function.In addition,it has been suggested that LCMT1 activity and expression might be regulated by changes in homocysteine metabolism[63].On the other hand,it has been described that phosphorylation of PP2AC at Thr304 or Tyr307 prevents the core enzyme to recruit the regulatory subunit(Figure 2),which is transient,disappearing within minutes[64-65].The transient deactivation of PP2A might enhance the transmission of cellular signals through kinase cascades within cells[66].The phosphorylation is reversed through an autophosphorylation mechanism[66].However,the identities of the kinases have remained obscure.For example,cyclin-dependent kinase 1(CDK1)has been implicated as a kinase capable of phosphorylating at Thr304 residue,reducing phosphatase activity[64].Additionally,the phosphorylation at Thr304 decreased B55 binding without affecting Leu309 methylation[59],and phosphorylation of the PP2AC at the Tyr307 site by receptor and nonreceptor protein tyrosine kinases can inactivate PP2A[65],including src kinase,epidermal growth factor receptor or insulin receptor[66].Other modifications of PP2AC include ubiquitination,which targets PP2A for degradation,and tyrosine nitration that increases PP2A activity in endothelial cells[57].
2.3 Noncanonical PP2A subunits Studies have suggested that monomeric PP2AC is unstable and rapidly degraded[50],thus existing atypical PP2A complexes with some other subunits including α4,TIPRL1,MID1,etc[67].α4 initially was shown to promote polyubiquitination and degradation of microtubule-associated PP2AC[68].However,subsequent studies revealed that α4 binds to PP2AC,which protects them from the proteasomal degradation until they are assembled into a functional phosphatase complex and promotes PP2A activation(Figure 2)[69-70].Several studies have demonstrated that α4 forms a stable complex directly with PP2AC via its structurally unordered,flexible C-terminus,independently of the A or B-type subunits,and α4-PP2ACinteraction domains not containing B regulatory subunits have identified[50,71].Furthermore,PP2AC-α4 interaction does not rely on PP2AC methylation[50].MID1 possesses E3 ligase activity which normally targets PP2AC for degradation through binding to its α4 regulatory subunit thereby stimulating proteasomal degradation of PP2AC and inhibiting the activity of PP2A(Figure 2)[68,72].However,some studies have reported that MID1 directly catalyzes the monoubiquitination and polyubiquitination of the α4,which triggers calpain-mediated cleavage(at the Phe255-Gly256 bond)of its MID1 binding domain and,ultimately,PP2AC polyubiquitination and proteasomal degradation[73-74].Besides,in mammalian cells,TIPRL1 is an inhibitory regulator of PP2A that has been shown to inhibit free PP2AC as well as PP2AD,and does not directly interact with α4 but rather primarily interacts with the PP2A subunits independently of mammalian target of rapamycin kinase activity,yielding an inactive phosphatase complex(Figure 2)[75-76],which demonstrate that TIPRL1 does not compete with α4 for PP2AC binding and that even a trimolecular TIPRL1-C-α4 complex can be formed and found as an endogenous complex in cells[76].Intriguingly,the A subunit was not detected in the isolated TIPRL1-C complex,thus suggesting that TIPRL1 may be part of both dimeric C-TIPRL1 and trimeric TIPRL1-C-α4 noncanonical PP2A complexes[50].

3 PP2A dysfunction and Alzheimer's disease
An increasing number of studies showed that PP2A activities are decreased in AD patient brains.Thus,PP2A dysfunction compromises the mechanisms controlling tau protein.However,the mechanisms underlying the downregulation of PP2A activity in AD are not completely understood and summarized in Figure 3.
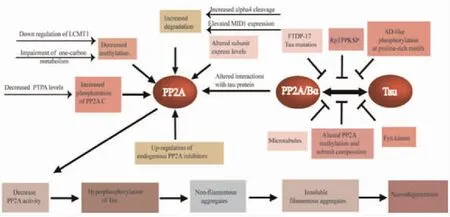
Fig 3 Overview of PP2A dysfunction in AD
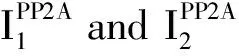
3.1 Alterations in PP2A Overall PP2A expression and activity are diminished in the cerebral cortex and hippocampus of AD.Deficits in PP2A activity are in line with the reported down-regulation of PP2AC at the gene[86],mRNA[87]and protein[88]expression levels in AD.There was a significant loss of regulatory B and B subunit mRNAs in the hippocampus of AD[87].Interestingly,cortical Bα regulatory subunit expression levels,but not the Bβ and B'α subunits were clearly decreased in AD[88].
3.2 Alterations in post-translational modifications Notably,the loss of neuronal PP2A holoenzymes regulatory subunit correlates with the imbalance of reversible PP2A methylation and severity of phosphorylated tau pathology in AD-affected brain[88-90].Accumulating evidence has shown that methylated PP2A(Leu309)is reduced while demethylated PP2A is increased,resulting in a reduction in the methyl/demethyl PP2A ratio of 75% in AD compared to controls[89,91],which may explain the decrease of PP2A activity and contribute to tau protein hyperphosphorylation[92].Significantly,the dramatic shift in PP2A carboxyl methylation appears to derive from an imbalance in the levels of the 2 highly specific PP2A modifying enzymes that control methylation,LCMT1,and PME-1.Of particular relevance,impacting PP2A carboxyl methylation and activity include elements of one-carbon metabolism such as levels of the methyl donor SAM,the competitive inhibitor,SAH,or the methylation cycle intermediate homocysteine[55,93].Some studies have shown that deficiency of B vitamins,such as folate,B12,and B6,leads to elevated homocysteine levels and an associated increased risk of cognitive impairment and AD[94-95],which increases SAH and diminishes SAM and glutathione S-transferase(GST)activity,thus leading to inhibition of SAM-dependent methylation reactions[96].Recently,emerging evidence has indicated that increased homocysteine increases the levels of phosphorylated tau protein as well as truncated tau protein species via caspase 3 activation,and enhanced tau protein oligomerization and aggregation by inactivating PP2A[97-98].Additionally,increased phosphorylation of PP2A at Tyr307 has been reported in post-mortem brain[99],likely mediated by PTPA.
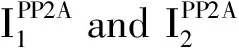
3.4 Disruption of PP2A-tau interaction Considerable evidence suggests that B55α-containing PP2A(PP2A/Bα)is by far the isoform with the strongest affinity for tau protein and most potent tau phosphatase activity in contrast to other PP2A holoenzymes[105-107].PP2A/Bα binds to the microtubule-binding domain of tau protein,and this interaction promotes tau protein dephosphorylation at multiple sites[107].Conversely,the PP2ADcore enzyme only weakly binds to tau protein and partially dephosphorylates tau protein without the Bα subunit[107].Several studies have indicated that the carboxyl methylation of PP2AC at Leu309 by LCMT1 is known to affect the association of B55α to form the B55α-PP2A holoenzyme[59,107-108].Therefore,decreased PP2A methylation levels are associated with disruption of PP2A-tau protein-protein interactions,thereby precluding PP2A-mediated tau protein dephosphorylation[109].As described above,disturbances in one-carbon metabolism result in reduced PP2A/Bα amounts in AD-affected brain.PP2A/Bα-tau protein-protein interaction can be inhibited by alterations in tau protein.AD-like pseudo-phosphorylation of several tau protein sites in the N-terminal proline-rich region of tau protein inhibits the association of tau protein with the dominant brain PP2A isoform PP2A/Bα[110].More remarkably,phosphorylation of Thr231 in the tau protein proline-rich230RTPPKSP236motif significantly decreases binding of tau protein to PP2A/Bα,providing a plausible explanation for the poor dephosphorylation of pThr231-tau protein by PP2A/Bα[111-112].Of note,phosphorylation of Thr231 occurs early in AD and inhibits further dephosphorylation of pathological pSer202/pThr205 tau sites by PP2A/Bα[111].Correspondingly,the synthetic RTPPKSP peptide competes with tau protein for binding to PP2A/Bα whereas the synthetic RpTPPKSP phosphopeptide failed to significantly inhibit PP2A/Bα-tau protein-protein interactions[112].In addition,Fyn binds in vitro to the230RTPPKSP236motif of tau protein,thereby inhibiting the interaction of PP2A with tau protein through competitive binding with PP2A/Bα[112].Interestingly,frontotemporal dementia with parkinsonism-17(FTDP-17)-associated missense mutations inhibit the binding of tau protein to PP2A/Bα by 20 to 70%[113].Surprisingly,the association of PP2A/Bα with microtubules induces an inhibitory effect on the tau phosphatase activity of PP2A,and PP2A/Bα activity is also inhibited by microtubule assembly dynamics[106].Besides,neurofilaments dynamics is regulated by PP2A activity.Therefore,neurofilaments may also indirectly interfere with PP2A/Bα-tau protein-protein interactions[114].However,it is not clear what is the primary mechanism of disrupting PP2A/Bα interactions.
4 PP2A-centric therapies for AD
Altogether,there is mounting evidence that PP2A dysfunction is a key player in the development of abnormally hyperphosphorylated tau protein pathology in AD.Consequently,there is a growing interest in developing PP2A-targeted therapies for AD,especially to counteract abnormally hyperphosphorylated tau protein pathology.Ideally,restoring neuronal methylation/phosphorylation of PP2AC using active compounds or dietary methylation donors and/or directly targeting the enzymes that control PP2A methylation,and/or targeting PP2A modulators could facilitate the assembly and viability of PP2A holoenzymes and increase PP2A-mediated dephosphorylation of tau protein.Many compounds and drugs currently used clinically have been known to modulate PP2A activity by several direct and indirect mechanisms[115],thereby facilitating tau protein dephosphorylation.
In this context,it is worth mentioning dietary compounds that target one-carbon metabolism and therefore influence PP2A methylation.Combined folate and vitamin B12 supplementation in aged rats with a chronically high level of homocysteine prevents hyperhomocysteinemia-mediated PP2A inhibition,tau protein hyperphosphorylation and memory deficits[116].Folic acid reduces tau protein phosphorylation by regulating PP2A methylation in streptozotocin-induced diabetic mice[117].It has been reported that dietary supplementation with SAM restored PP2A activity,and GST activity because SAM is an essential cofactor for GST[118].Additionally,morroniside(MOR), isolated fromCornusofficinalis,was found to induce PP2A activation by inhibiting phosphorylation of PP2AC at Tyr307,resulting from decreased Src activity through dephosphorylation at Tyr416[119].On the other hand,there are also efforts to target the enzymes that control PP2A methylation,LCMT1,and PME-1.For instance,MOR may promote methylation of PP2A at Leu309 via decreasing PME-1 expression[119]; and eicosanoyl-5-hydroxytryptamide(EHT),a naturally occurring component from coffee beans,increase PP2A activity by inhibiting PME-1[120-121].Administration of SIG-1012 has been shown to lower tau protein phosphorylation by blocking PME-1 activity in rats[122].Additionally,pseudoginsenoside-F11 enhanced the activity of PP2A by increasing the protein levels of LCMT-1,thus alleviating cognitive deficits[123].
PP2A is also targeted by numerous endogenous regulators effecting PP2A viability.For instance,memantine,an anti-AD drug,inhibits this pathology by directly targeting SET[124],and COG112,a SET interacting peptide,interacted with SET protein and inhibited the association between SET and PP2AC[125].Additionally,metformin,an anti-diabetic drug,activate PP2A activity via interfering with the association of the PP2AC to the MID1-α4 protein complex[126].Resveratrol also increases PP2A activity and dephosphorylates tau protein by interfering with the MID1-α4-PP2A complex and reducing the MID1 transcript and protein level[72].Remarkably,multiple lines of evidence have indicated that supplementation of less toxic sodium selenate improves spatial learning and memory and mitigates motor deficits in tauopathy transgenic mouse models,which significantly boosts PP2A activity[127-128].Together,these may be developed as new drugs for AD treatment after further investigation.Besides,other therapeutic strategies such as inhibition of AEP,restoration of PTPA,targeting PP2A-tau protein-protein interactions,etc.may be therapeutically useful for treating tau protein-mediated neurodegenerative diseases[58].
5 Conclusions
Initially,potential anti-tau therapies were based mainly on inhibition of kinases or tau protein aggregation,or on stabilization of microtubules,or Aβ-targeting treatments,but most of these approaches have been discontinued because of toxicity and/or lack of efficacy[129].Currently,targeting the major tau protein phosphatase PP2A is an attractive strategy to counteract abnormally hyperphosphorylated tau protein pathology.However,the approach of simply activating PP2A-dependent tau protein dephosphorylation is likely flawed[57].Firstly,regulation of PP2A in AD is multidimensional and intrinsically complex.Moreover,it is not known which mechanism leads primarily to PP2A dysfunction.For instance,besides the complexity of PP2A regulation mentioned above,PP2A and glycogen synthase kinase-3β(GSK-3β)affect the activities of each other.A growing body of evidence has indicated that GSK-3β regulates PP2AC methylation by suppressing the expression of PME-1 and phosphorylating LCMT-1 and that PP2A can upregulate GSK-3β activity via dephosphorylation at Ser9 in a Leu309 methylation-independent manner[130-131].Thus,in AD patients,dysregulations of GSK-3β and PP2A impact tau protein phosphorylation directly and indirectly through affecting the activities of each other.Herein,more compounds,other than a single compound,are likely needed to overcome the many-sided PP2A deficits that are present in AD.Secondly,specificity and side-effects of PP2A activators are likely to be a huge issue,due to the broad spectrum of PP2A enzymes and complex PP2A regulatory mechanism.Although PP2A/Bα is the primary tau protein phosphatase in the human brain,none of the compounds tested so far have been demonstrated isoform specificity.In addition,drugs may also target PP2A enzymes outside the brain and in brain regions not primarily involved in the disease process.Furthermore,There is also considerable evidence have shown that pathological brain changes manifest at least one decade before the appearance of AD symptoms[6,132]; so it is likely that a major challenge will be the toxicity of long-term use of PP2A/tau protein targeting compounds,due to the treatment need of extended pathological course of AD.Thus,treatment with pharmaceutical drugs may require to be started early,in particular,with the future improvements in diagnostics.Lastly,the observation that PP2A targeting compounds can improve AD-like pathology derives from testing in mouse models that do not reproduce the complexity of the pathology encountered in their human counterparts.While promising,the validity of these PP2A/tau protein targeting compounds or drugs for AD needs to be further investigated.
Based on these data and conclusions mentioned throughout the review,a better understanding of the role of PP2A and its modulators in the regulation and cellular functions is paramount on the challenging and tangled path to guaranteeing the success of novel PP2A-centric therapeutic strategies and to development of effective drugs for AD.Accordingly,a future potential therapy for AD could be based on agents that increase the activity of specific phosphatases; and the development of several compounds interfering with PP2A status will pave new avenues to the development of rational therapeutic intervention strategies aimed at the prevention and/or treatment of AD.



