李媛,马小雪
家族性低磷血症性佝偻病(FHR)是罕见的遗传性佝偻病,由一组以肾脏磷酸盐运输缺陷导致排泄磷酸盐过多和低血磷为特征的疾病组成,患者1,25二羟基维生素D〔1,25(OH)2D〕降低或处于参考范围内[1]。FHR的临床特征为生长迟缓、儿童期佝偻病、骨软化、牙齿发育不良。迄今,已确定了6种遗传性佝偻病,包括常见的X-连锁显性遗传佝偻病(XLH,MIM307800)、偶见的常染色体显性遗传低磷性佝偻病(ADHR)、常染色体隐性遗传低磷性佝偻病(ARHR1,MIM 241520;ARHR2,MIM613312;ARHR3,MIM259775)、 罕见的X-连锁隐性遗传佝偻病(XRHR)、罕见的低磷性佝偻病伴高钙尿(HHRH,MIM241530)、低磷性佝偻病伴甲状旁腺功能亢进(MIM612089)[2]。XLH的致病基因为X染色体内肽酶同源性的磷酸调节基因(PHEX基因),而成纤维生长因子23(FGF23)突变可引起ADHRD,牙基质蛋白1(DMP1)、核苷酸外焦磷酸酶/磷酸二脂酶1(ENPP1)、序列相似家族成员20(FAM20C)分别为ARHR1、ARHR2、ARHR3的致病基因[3],XRHR的致病基因为CLCN5,钠磷共转运蛋Ⅱ型溶质转运家族34(SLC34A3)突变可引起HHRH[4-5]。其中XLH最为常见,发病率约为1/20 000[6]。
FHR临床表现多样,严重程度不一,给临床诊断带来很大困难。因此,尽早地明确基因诊断对临床诊治、了解疾病预后至关重要。本文对1例患有FHR的汉族患儿采用二代测序技术进行相关基因检测,发现PHEX基因的新突变:c.2066C>T(p.Ala689Val);再用Sanger测序法验证患儿及其主要家族成员,发现该家系中患者均存在同样的基因突变,确诊该家系为PHEX基因新突变致XLH,现报道如下。
1 病例简介
先证者,女,1岁7个月,汉族,因出现“O”型腿半年于2015年5月至云南省第一人民医院就诊。半年前患儿家长发现其双小腿弯曲,外院诊断为“佝偻病”,服用碳酸钙1片/d,无效。患儿自出生后1个月起每天服维生素AD滴剂,10个月出牙。查体:身长83 cm,体质量12 kg,前囟闭合,出牙12颗,轻度双膝内翻,双膝间距2.5 cm。下肢X线检查未见佝偻病表现(见图1),肾脏B超正常。家系调查:父母非近亲结婚,父亲、祖母、大姑妈均有身材矮小、重度双膝内翻。患儿及其主要家庭成员实验室检查资料见表1,家族系谱见图2,初步诊断为FHR。根据家族系谱,临床考虑XLH,但不能排除ADHR,为明确诊断进行基因测序。患儿家长签署知情同意书后,抽取先证者及其主要家族成员静脉血2.5~3.0 ml置于EDTA抗凝管中,送昆明金域医学检验所进行基因测序。采用二代测序法(NGS)对先证者进行FHR相关基因测序,发现PHEX基因突变,再用Sanger测序法对先证者及其主要家族成员PHEX基因突变进行验证,将测序发现的突变在ESP6500、dbSNP、千人基因组数据库进行比对查阅。
本文价值:
家族性低磷血症性佝偻病(FHR)是罕见的遗传性佝偻病,由一组肾脏排泄磷酸盐增多导致低血磷的疾病组成,共同的临床特征为生长迟缓、儿童期佝偻病、骨软化、牙齿发育不良。本文对1个FHR家系的临床表现、生化指标及影像学资料进行研究,通过基因测序发现该家系中4例患者均有PHEX基因的新致病突变(c.2066C>T),从而确诊。本研究丰富了人类PHEX基因突变数据库的种类,有助于深入了解该基因的结构与功能。
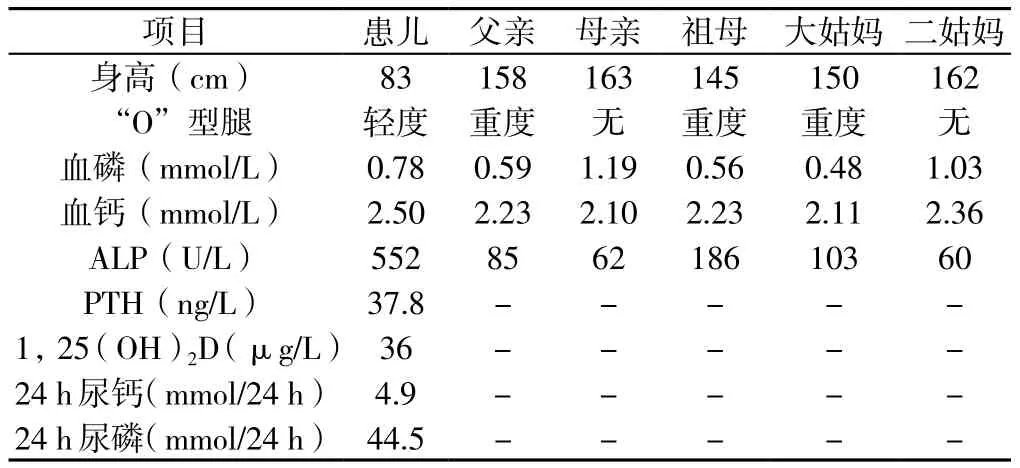
表1 FHR患儿及其主要家族成员实验室检查资料Table 1 Laboratory examinations of the children and major family members of the FHR
基因测序发现先证者(Ⅳ:3)PHEX基因存在c.2066C>T(p.Ala689Val)杂合子突变。经家系验证分析,家族中患者(Ⅱ:1、Ⅲ:1、Ⅲ:5)均检测到相同基因位点突变,而未受影响的家族成员中则无基因突变(见图3),提示此变异为致病突变。该突变为错义突变,位于20号外显子,突变导致第689位丙氨酸为缬氨酸所取代,蛋白多肽链原有功能丧失。该突变未见文献报道,ESP6500、dbSNP、千人基因组数据库中均未收录,为新的致病突变。该家系中,3个女性患者为PHEX基因杂合子突变,1个男性患者为PHEX基因半合子突变,XLH诊断明确。
确诊后予患儿口服磷酸盐(元素磷剂量为20 mg·kg-1·d-1)、骨化三醇20 ng·kg-1·d-1。2个月后复查,血磷1.08 mmol/L、血钙2.5 mmol/L、碱性磷酸酶(ALP)432 U/L、尿磷29.5 mmol/24 h、尿钙4.9 mmol/24 h、尿肌酐6 630 μmol/L、甲状旁腺激素(PTH)35.1 ng/L。后每3个月进行1次门诊随访,血磷0.82~1.04 mmol/L、ALP 368~397 U/L,尿钙/尿肌酐<0.4。治疗1年后下肢X线检查示“O”型腿减轻(见图4)。患儿3岁半时身高99.6 cm,下肢 “O”型腿已不明显,肾脏B超正常。
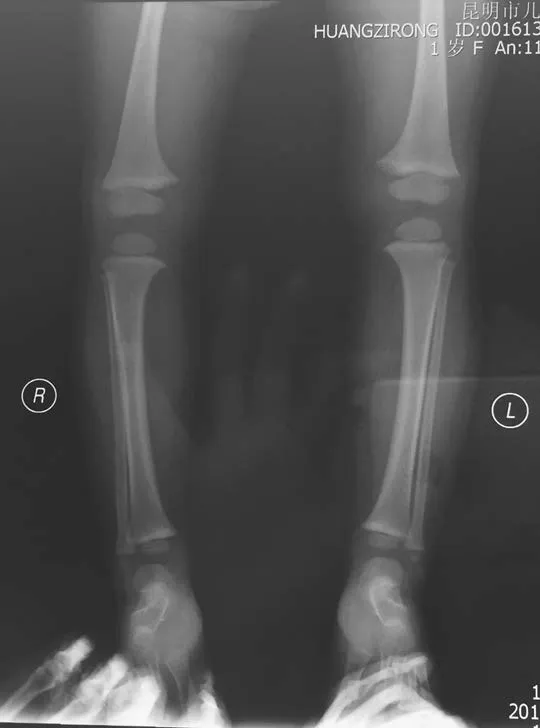
图1 先证者下肢X线检查结果Figure 1 X-ray image of the lower extremity of the proband
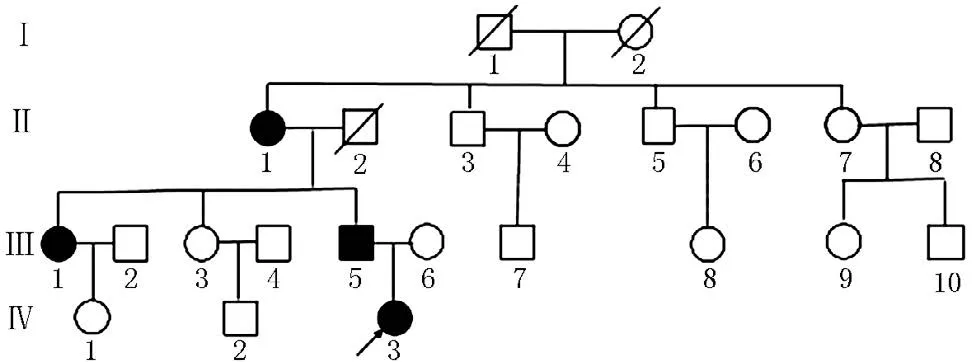
图2 FHR家族系谱Figure 2 Pedigree of the FHR
2 讨论
FHR是一种罕见的遗传疾病,临床表现多样。XLH是最常见的遗传性佝偻病,86%~87%的FHR为XLH[5]。XLH由FGF23介导的低血磷所致,主要临床特征为身材矮小且不成比例、佝偻病、下肢畸形,患者还可能出现牙齿及牙龈异常、肌肉无力等表现[4]。XLH的生化特征包括低血磷,1,25(OH)2D低或处于参考范围, 成纤维细胞生长因子23(FGF23)升高,尿磷排泄增多,ALP升高(儿童)或处于参考范围(成人),PTH升高或处于参考范围,血钙处于参考范围,尿钙排泄降低或处于参考范围[1,5]。
本文针对1个遗传性佝偻病家族进行分析,该家族中患者均有相似的特征:身材矮小(除先证者外)、“O”型腿、低血磷,而血钙处于参考范围,男女患者临床表现无明显差别。根据家族系谱,临床考虑XLH,但不能排除ADHR。因此,采用NGS对先证者(Ⅳ:3)进行遗传性佝偻病基因测序,发现PHEX基因存在c.2066C>T(p.Ala689Val)杂合子突变。再用Sanger测序法对先证者及其主要家族成员PHEX基因突变进行验证,显示家族中患者(Ⅱ:1、Ⅲ:1、Ⅲ:5)均存在相同基因位点突变,而未受影响的家族成员则不存在基因突变,提示此变异为致病突变。该突变未见文献报道,ESP6500、dbSNP和千人基因组数据库中均未收录,因此确定为新的致病性突变。
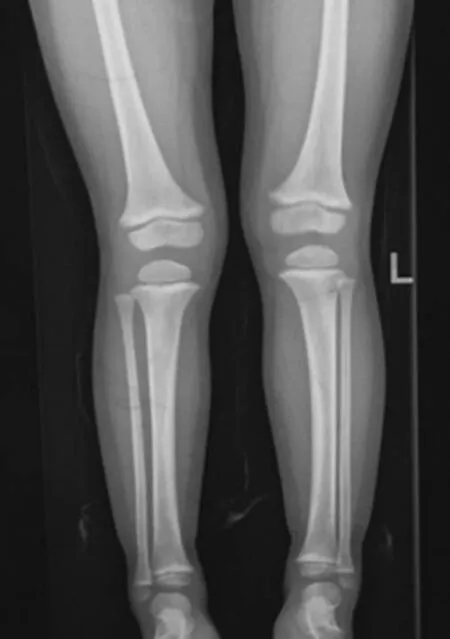
图4 治疗1年后先证者下肢X线检查结果Figure 4 X-ray image of the lower extremity of the proband after 1-year treatment
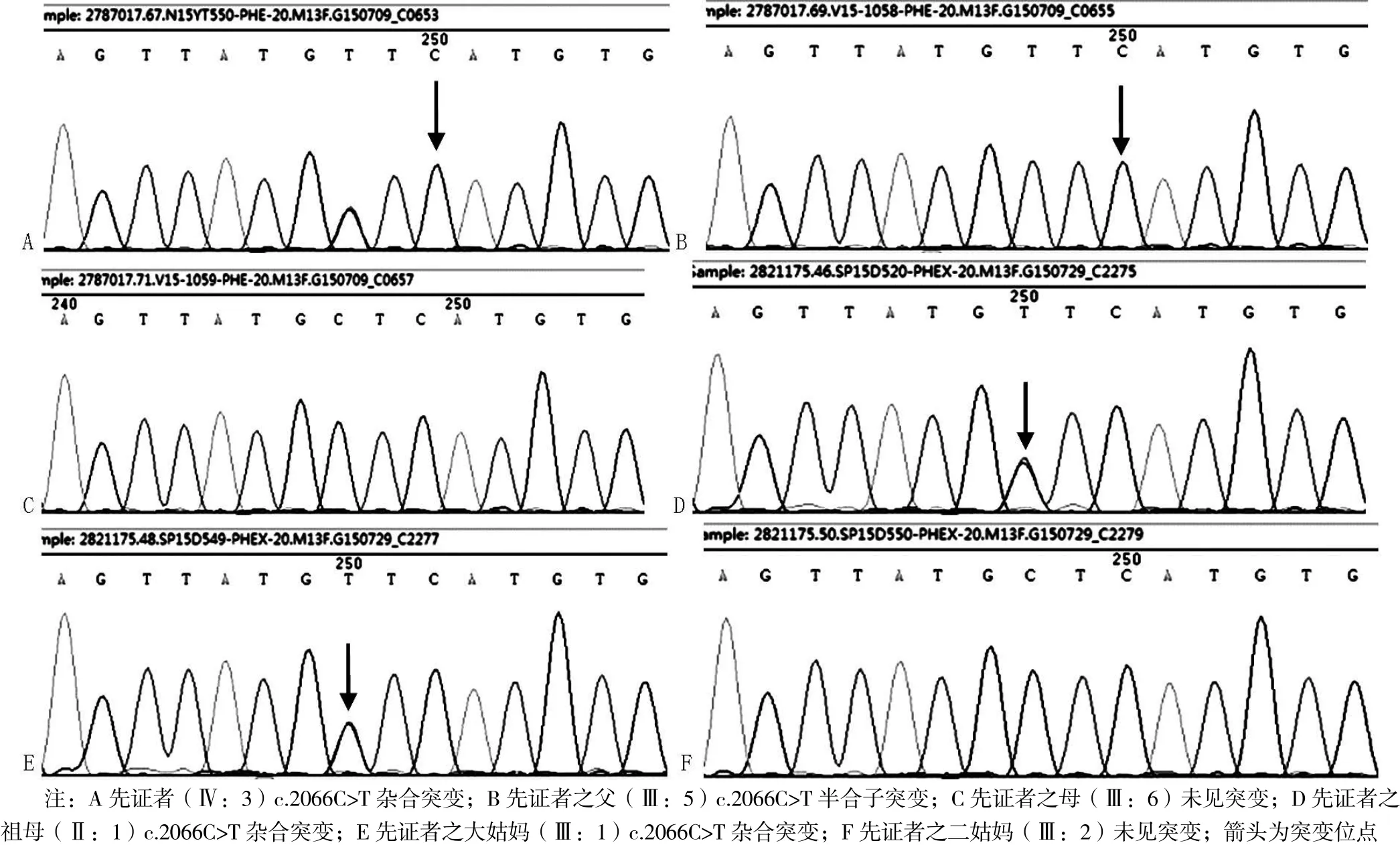
图3 X染色体内肽酶同源性的磷酸调节基因测序图Figure 3 Sequence diagram of the PHEX gene
XLH的致病基因PHEX定位于染色体XP22.1,由22个外显子组成,编码1条由749个氨基酸构成的蛋白[7]。PHEX由1个小的氨基末端的胞内尾巴、1个短的跨膜域和一个大的羧基末端细胞外域组成[1],后者包含10个高度保守的半胱氨酸残基,2个锌指结构域,与二级结构或催化活性有关[8]。PHEX主要在骨和牙齿中表达[9]。PHEX的功能丧失伴随骨骼矿化缺陷、FGF23的合成和分泌增强。FGF23增高不仅导致肾脏磷酸盐重吸收减少,而且抑制肾脏1-α羟化酶的活性,使得25羟基维生素D〔25-(OH)D〕生成减少[4]。然而,PHEX并不直接分解FGF23,PHEX的功能仍然有待阐明。
迄今,全世界报道的XLH患者PHEX基因突变共338种,其中移码突变占25%,异常剪切突变占23%,错义突变占22%,无义突变占18%,缺失占8%,还有4%基因多态性[10]。这些突变分散于PHEX基因,无明显突变热点[11-12]。20号外显子的突变共19种,其中移码突变9种,无义突变4种,错义突变4种,缺失2种。目前未发现基因突变类型、突变位置与临床严重程度之间的关联[7,11-13]。
本文发现的PHEX基因新突变c.2066C>T(p.Ala689Val)为错义突变,突变位于20号外显子,导致第689位丙氨酸为缬氨酸所取代,突变点下游的阅读框漂移改变了编码产物的性质,造成PHEX功能丧失。XLH为完全外显,理论上由于X染色体的随机失活,杂合子突变的女性患者病情轻,而半合子突变男性则病情较重。但在本文中男、女患者临床表现无明显差别,与X染色体失活倾斜阻止了正常基因的表达有关[2],而先证者病情较轻可能与X染色体随机失活的程度较轻有关[2]。
目前XLH的治疗方法是口服骨化三醇和磷酸盐合剂。美国XLH临床诊治指南推荐骨化三醇的剂量为20~30 ng·kg-1·d-1,分 2~3次服用;磷酸盐剂量为 20~40 mg·kg-1·d-1(最多为2~3 g/d),分3~5次服用[13]。治疗过程中需每3个月监测血钙、磷、肌酐及尿钙/尿肌酐和PTH。尿钙/尿肌酐>0.4,提示骨化三醇剂量过大,应及时减量[7];PTH升高可通过增加骨化三醇的剂量或减少磷酸盐剂量来纠正[14]。由于血磷波动较大,不建议用血磷衡量磷的需要量[7]。而应依据身高和生长速度及骨骼畸形改善情况判断药物剂量是否符合要求[14]。本研究中,先证者病情较轻,骨化三醇、磷酸盐起始剂量分别为20 ng·kg-1·d-1、20 mg·kg-1·d-1,监测血钙、血磷、血肌酐、PTH及尿钙、尿肌酐,根据尿钙/尿肌酐调整骨化三醇的剂量,磷酸盐的剂量则以达到空腹血磷处于参考范围下限即可。本例患儿身高年增长速率为8.6 cm/年,且下肢畸形逐渐消失,说明药物剂量适宜。
国外研究显示,在出现生长障碍及佝偻病体征前的婴儿期开始治疗的XLH患者,在身高改善及病情减轻方面较1岁后开始治疗者好[15]。XLH患者的子女应在出生后3个月内仔细追踪,尽早治疗。在婴儿期诊断XLH,除阳性家族史外,还基于低血磷、高ALP,而低血磷及高ALP通常在生后3个月内出现[15]。本文中先证者在外院被误诊为“佝偻病”治疗半年,误诊的原因除医生对本病认识不足外,更与不重视家族史及血生化检查有关。所幸患儿病情较轻,确诊时年龄不大,治疗效果好。
综上所述,本文确定了1例FHR家系PHEX基因的新致病突变,丰富了人类PHEX基因突变数据库的种类,有助于深入了解该基因的结构与功能。本文通过基因检测确诊患者为XLH,对先证者进行及时治疗并取得了满意疗效,同时也为该家系的遗传咨询和今后的产前诊断提供了可靠的依据。
本文局限性:
因检测条件限制,本研究未检测该家系患者血成纤维生长因子23(FGF23)水平,不能了解PHEX基因的新致病突变(c.2066C>T)对血FGF23水平的影响。此外,如能在先证者的随访中监测血甲状旁腺激素(PTH)水平的变化,更有助于骨化三醇和磷酸盐剂量的调整。
作者贡献:李媛进行文章的构思与设计,撰写论文,负责文章的质量控制及修改、审校,对文章整体负责;马小雪参与研究的实施、数据收集。
本文无利益冲突。



