闫露露,楼碧莹,徐文蓉,李海波
中国人群腺苷脱氨酶2缺乏症致病基因携带者筛查
闫露露1,楼碧莹2,徐文蓉3,李海波1
1.宁波市妇女儿童医院出生缺陷综合防治实验室,浙江宁波 315000;2.宁波大学医学部,浙江宁波 315000;3.北京智因东方转化医学研究中心有限公司,北京 100023
分析中国人群腺苷脱氨酶2缺乏症(deficiency of adenosine deaminase 2,DADA2)致病基因频率,为有效开展中国人群DADA2的防控和干预工作提供依据。回顾性分析2015年1月至2021年1月于北京智因东方转化医学研究中心有限公司进行全外显子组测序的11.1万例原始数据,对基因致病和可能致病性突变进行携带者筛查,分析基因杂合突变的类型和等位基因频率。发现29种基因致病和可能致病性杂合突变,其中6种[c.730G>T(p.E244X)、c.1049_c.1061delAGCTGCCTTACTT(p.K350Tfs*14)、c.940A>T(p.K314X)、c.1239+2(IVS8)T>C、c.1240-1(IVS8)G>A、c.1087C>T(p.Q363X)]既往未见报道,等位基因累计频率为0.58%。人群基因频率最高的为p.F355L(0.5%),其次为p.Y453C(0.014%)和p.P193L(0.013%),其他26种位点均小于0.01%,为罕见突变位点。通过筛查丰富了基因突变谱,明确了中国地区人群的基因突变的携带率约1/171,推测中国人群的DADA2患病率约1/116 964。
腺苷脱氨酶2缺乏症;基因;等位基因频率;患病率
腺苷脱氨酶2缺乏症(deficiency of adenosine deaminase 2,DADA2)是一种罕见的常染色体隐性自身炎症性疾病,由基因纯合或复合杂合突变导致,临床表现为自身炎症反应综合征、卒中反复发作、类似结节性动脉炎的血管炎及免疫缺陷等[1-2]。迄今累计报道病例约300多例[3]。Infevers数据库目前共收录104种基因致病和可能致病性突变,其中大多突变类型为错义突变、无义突变和剪接突变。目前DADA2的治疗主要是使用肿瘤坏死因子抑制剂(tumor necrosis factor inhibitor,TNFi)。一旦确诊,应尽早应用TNFi以减轻或避免神经系统损伤。对血液病或TNFi难治性患者建议应用造血干细胞移植[4-6]。有研究报道DADA2的患病率约1/222000[7]。但我国人群DADA2的研究报道甚少,临床上对其认识有限,患者可能因未及时诊断错过最佳治疗时机。因此,本研究基于中国人群大样本的全外显子组测序(whole exome sequencing,WES)数据进行回顾性分析,以了解中国人群DADA2的患病率和致病基因突变携带情况,为制订有效可行的DADA2防控策略提供依据。
1 资料与方法
1.1 研究对象
回顾性分析2015年1月至2021年1月北京智因东方转化医学研究中心有限公司的11.1万例WES原始数据,筛查内容及范围为基因致病和可能致病性突变位点,排除临床意义未明或良性突变位点。本研究经宁波市妇女儿童医院伦理委员会审查通过(伦理审批号:[2019]伦审字(41)号)。
1.2 研究方法
Fastq格式的原始数据进行fastp(0.23.1)质控,去除接头、低质量reads等处理后,以GRCH37/hg19基因组版本为依据进行突变检测及注释。利用BWA0.7.17软件将测序序列与GRCH37/hg19基因组版本比对,使用GATK3.70软件获取单核苷酸多态性(single nucleotide polymorphism,SNP)和插入缺失。利用ANNOVAR和Vep软件进行突变注释,根据突变的类型、普通人群频率注释(如ExAC、ESP、GnomAD等)及HGMD、OMIM、ClinVar等数据库的关联注释,筛选出致病和可能致病性突变。
1.3 统计学方法
某种突变的阳性检出例数除以所有入组人群数量,得出该突变的人群等位基因频率。使用哈迪–温伯格平衡检验,估计疾病的患病率。
2 结果
2.1 ADA2基因携带情况分析
在11.1万例WES数据中,共检出29种基因致病和可能致病性杂合突变,等位基因累计频率为0.58%,各筛查位点的携带情况见表1。其中错义突变占比最高,达55.17%(16/29);其次为无义突变,占24.14%(7/29);移码突变占13.79%(4/29),剪接突变占6.90%(2/29)。等位基因频率位于前十的突变分别为c.1065C>A(p.F355L)、c.1358A>G(p.Y453C)、c.578C>T(p.P193L)、c.97_c.98insA(p.T33Nfs*29)、c.139G>C(p.G47R)、c.916C>T(p.R306X)、c.1069G>A(p.A357T)、c.278T>C(p.I93T)、c.25C>T(p.R9W)和c.139G>T(p.G47W),其中等位基因频率最高的突变为c.1065C>A(p.F355L),达0.5%,其次为c.1358A>G(p.Y453C)(0.014%),c.578C>T(p.P193L)(0.013%),上述3个突变携带者人数占所有杂合携带者人数的78.81%(372/472)。其余26种突变均为罕见突变,等位基因频率远小于0.01%。
2.2 ADA2基因突变分布情况
29种基因致病和可能致病突变中,有2种为剪接突变,位于8号内含子,其余27种均位于不同的外显子上。后者中70.37%位于ADA2蛋白的催化结构域,p.M1T和p.R9W位于ADA2蛋白的N端信号肽,p.R45W、p.G47V、p.G47W、p.G47R位于ADA2蛋白的二聚结构域,但本研究并未发现位于PRB结构域上的相关突变,见图1。
3 讨论
DADA2是一种罕见的常染色体隐性遗传病,于2014年由Zhou等[1]首次报道。该病典型的临床特征包括发热、结节性动脉炎、网状青斑、早发型卒中和免疫缺陷。DADA2的临床异质性很大,患者通常在儿童期发病,从发现到确诊往往需要很长的时间。目前国内已报道41例DADA2患者,年龄范围为9天至25.6岁,平均诊断年龄7.25岁,该病临床表现多样,反复发热和皮肤受累是DADA2最常见的特征[6,8-13]。41例患者中有37例存在反复发热,87.80%的患者存在皮肤受累,皮肤表现最常见的特征为结节红斑病、网状青斑、口腔溃疡和手指坏疽,高于国外报道,可能与种族差异及国内目前报道病例较少有关[1]。神经系统病变是DADA2患者的另一个主要特征,41例患者中有25例出现缺血性或出血性卒中。其他常见特征包括关节炎/关节痛、肝脾大。其他不常见的特征包括心肌炎、肌肉无力和肌炎。41例患者中有3例因感染或器官衰竭死亡,死亡率7.32%,与其他报道相似[14-15]。国内DADA2患者绝大部分在10岁前发病,多数患者存在不同程度的器官受累,因此,对儿童期出现发热、早发性卒中反复发作、系统性血管炎、免疫缺陷等病变的患儿应考虑DADA2,尽早进行血浆ADA2酶活性和基因检测。
基因位于染色体22q11.1,包含10个外显子,编码一种由髓细胞分泌的细胞外酶,主要在活化的单核细胞、巨噬细胞和树突细胞上表达[1-2]。ADA2是一种具有四个结构域的二聚酶,包括N端的信号肽、二聚结构域、催化结构域和推定受体结合结构域[16]。研究表明缺乏可导致血管内皮损伤,破坏血管壁完整性,导致炎症和血管损伤[1,17]。DADA2是为数不多的可治疗的自身免疫性疾病,对中国人群DADA2患病率的评估暂无文献报道,因此对基因致病突变携带者的筛查尤为重要,可通过了解人群基因的等位基因频率,评估人群中DADA2的发病率,进而为DADA2的诊断防治提供理论依据。
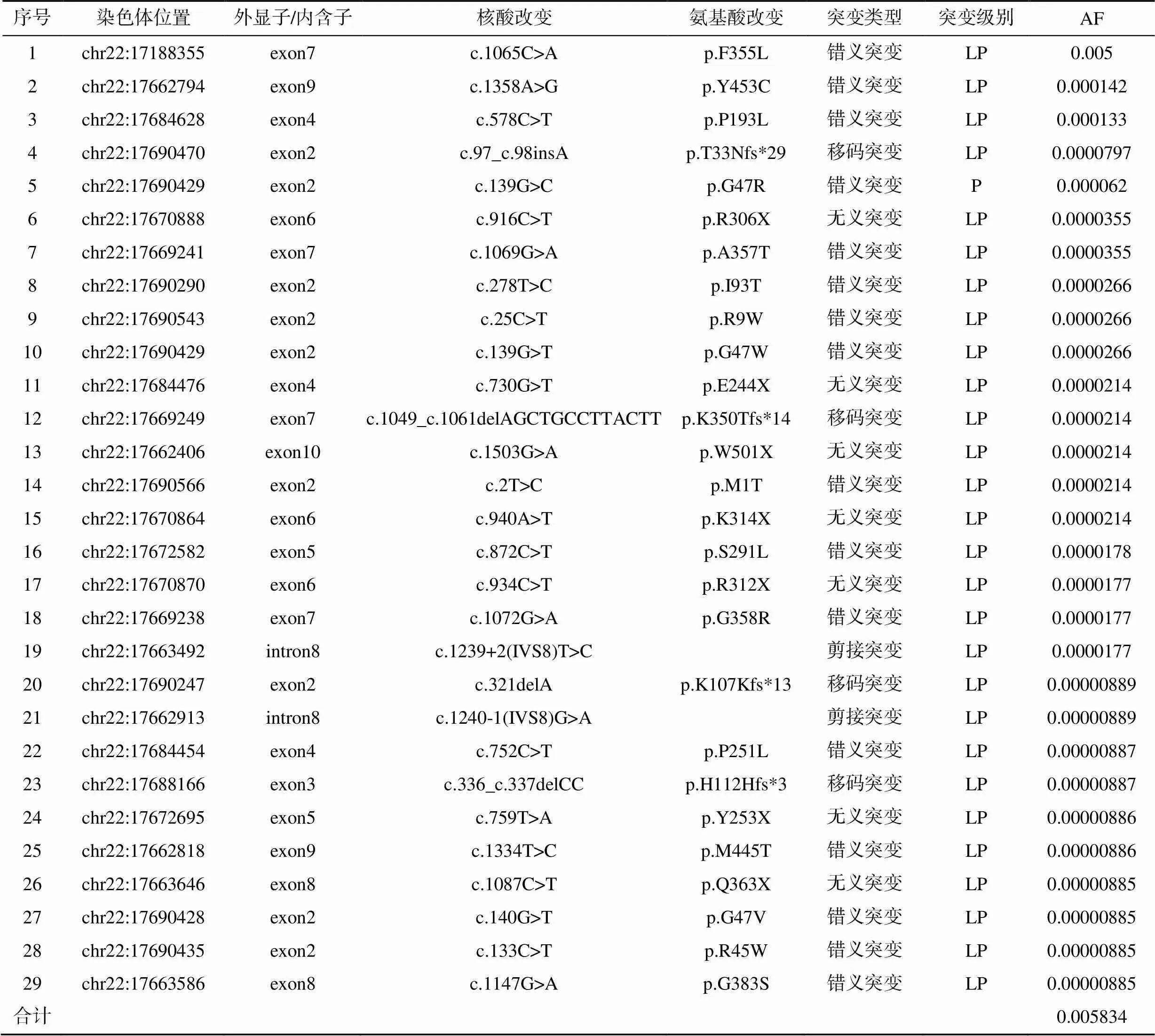
表1 11.1万例WES数据分析ADA2基因突变携带情况
注:AF为等位基因频率;P为致病的;LP为可能致病的
注:红色为未见报道的新突变
目前发现的基因致病突变种类多样,均分布在10个外显子中。检索ClinVar和Infevers数据库,共发现120个基因可能致病和致病性突变,其中主要为错义突变,占60%(72/120),移码突变占17.5%(21/120),无义突变占8.3%(10/120)。已报道的DADA2患者多为复合杂合突变。研究表明p.G47R、p.G47A、p.R169Q和p.Y453C等突变为最常见的致病位点[1-2]。这些突变可能改变ADA2蛋白受体结合域,影响蛋白的二聚化、催化活性和分泌,并进一步诱发DADA2的疾病表型。p.G47R纯合突变常见于格鲁吉亚‑犹太和土耳其人群,该突变在格鲁吉亚–犹太人群的携带率为10.2%,在土耳其人群为2%[1-2,18]。p.R169Q主要见于芬兰、荷兰和比利时人群,其在芬兰人群的携带率最高,为1/160,患病率约1/103 217;p.T360在意大利人群中较为常见,而p.Y453C则是最常见的泛民族突变体[19]。尽管在格鲁吉亚–犹太人群中已经发现部分DADA2患者,但考虑到p.G47R在该人群中高比例的携带率,很可能有一部分患者未得到及时的诊断。
本研究结果显示基因致病突变在中国人群的累计等位基因频率为0.58%,共发现29种基因致病突变。其中仅p.F355L的等位基因频率达到0.5%,该位点可能为中国人群的突变热点,且该突变在东亚人群中的基因频率为0.28%。Hashem等[4]曾在一名DADA2患者中发现p.F355L/p.E328K复合杂合突变。p.Y453C在中国人群等位基因频率为0.0142%,该突变在国外比较常见;Caorsi等[16]报道过3例DADA2患儿均为基因复合杂合突变,表现出DADA2特征性的临床表现,包括发热、网状青斑、出血性卒中、肝大和全血细胞减少等。青岛发现一例p.Y453C纯合突变患儿,临床表现为反复发热、脾大和脂膜炎,但无血液系统和神经系统异常[13]。p.P193L在中国人群的基因频率为0.013%,Belot等[20]在一个法国DADA2男性患者中发现基因p.P193L/ p.R169Q复合杂合突变,患者表现为反复发热并伴有口腔溃疡和关节疼痛、结节性多动脉炎、缺血性卒中和颈内动脉狭窄等临床特征,其哥哥28岁时死亡,表现为终生炎症特征,包括腿部活动性病变和四肢坏死等。c.97_c.98insA(p.T33Nfs*29)为移码突变,在中国人群的等位基因频率为0.008%,该突变暂无文献报道。c.139G>C(p.G47R)在中国人群的基因频率为0.006%,有多篇文献报道在不同种族中发现p.G47R纯合和复合杂合突变[2,21-22]。
p.R306X为无义突变,其在中国人群等位基因频率为0.0036%,Garg等[21]曾报道1例患有严重血管疾病的5岁土耳其女孩携带p.R306X/p.G47R复合杂合突变。c.1069G>A(p.A357T)在中国人群等位基因频率为0.0036%,Poswar等[22]报道1例p.A357T纯合突变的12岁患者,表现为反复发热、溃疡、网状青斑、中脑梗死和椎动脉瘤等。c.278T>C(p.I93T)在中国人群等位基因频率为0.0027%,2014年Zhou首次在一名15岁的英国男孩中发现p.Ile93Thr/p.Met1Thr复合杂合突变,患儿有反复发热、腔隙性脑梗死和皮肤表现,最终死于并发症[1]。c.25C>T(p.R9W)在中国人群等位基因频率为0.0027%,有文献报道1例携带p.R9W/p.Gl47A复合杂合突变,表现为发热、疼痛的皮下结节、脑膜炎和脑干梗死等[22]。p.G47W在中国人群等位基因频率为0.0027%,Barron等[23]曾报道1例携带G47W/R169Q复合杂合的DADA2患儿,临床表现为中性粒细胞减少、菌血症,急性移植物抗宿主病。此外,本研究发现6种国内外暂无文献报道的基因杂合突变,即c.730G>T(p.E244X)、c.1049_c.1061delAGCTGCCTTACTT(p.K350Tfs*14)、c.940A>T(p.K314X)、c.1239+2(IVS8)T>C、c.1240-1(IVS8)G>A、c.1087C>T(p.Q363X),上述突变的等位基因频率均远小于0.01%,为罕见突变位点,丰富了基因突变谱。
单基因疾病的患病率可通过人群中报告的有害突变的总等位基因频率来估计,有研究通过人群WES数据估计疾病的患病率[24]。因此,笔者通过中国人群的大样本的WES数据分析中国人群致病基因频率可获得更精准的DADA2患病率。根据本文分析结果,推测我国人群基因的携带率约1/171,DADA2的患病率约1/116 964,比Jee等[7]通过整合gnomAD数据库(超过14万人的基因组数据)中突变的等位基因频率和致病突变的功能数据得出的患病率要高,可能原因是样本量和种族差异所致。中国人口近14亿,理论上DADA2患者约11 969人,考虑到DADA2外显不全和表现度差异性等,有相当数量的DADA2患者仍未被及时诊断,严重阻碍DADA2防治能力的提升。本文通过筛查中国人群的致病等位基因频率情况,估计的DADA2发病率可为制订中国人群DADA2的预防计划提供有价值的参考。
[1] ZHOU Q, YANG D, OMBRELL A K, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2[J]. N Engl J Med, 2014, 370(10): 911–920.
[2] NAVON E P, PIERCE S B, SEGEL R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy[J]. N Engl J Med, 2014, 370(10): 921–931.
[3] ESCHERICH C, BOTTICHER B, HARMSEN S, et al. The growing spectrum of DADA2 manifestations- diagnostic and therapeutic challenges revisited[J]. Front Pediatr, 2022, 10: 885893.
[4] HASHEM H, KELLY S J, GANSON N J, et al. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders[J]. Curr Rheumatol Rep, 2017, 19(11): 70.
[5] PINTO B, DEO P, SHARMA S, et al. Expanding spectrum of DADA2: A review of phenotypes, genetics, pathogenesis and treatment[J]. Clin Rheumatol, 2021, 40(10): 3883–3896.
[6] LI G M, HAN X, WU Y, et al. A cohort study on deficiency of ADA2 from China[J]. J Clin Immunol, 2023, 43(4): 835–845.
[7] JEE H, HUANG Z, BAXTER S, et al. Comprehensive analysis of ADA2 genetic variants and estimation of carrier frequency driven by a function-based approach[J]. J Allergy Clin Immunol, 2022, 149(1): 379–387.
[8] ZHAO X, ZHANG J, LI C, et al. Early onset is an indication of the severity of DADA2 disease[J]. Rheumatology (Oxford), 2023, 62(2): 969–976.
[9] 李冀, 武跃芳, 钟林庆, 等. 儿童腺苷脱氨酶2缺乏症1例临床特征和基因分析并文献复习[J]. 中国实用儿科杂志, 2018, 33(10): 800–804.
[10] 宋晓晨, 周芳. 清髓性造血干细胞移植治疗CECR1基因突变致腺苷脱氨酶2缺乏症一例[J]. 中华儿科杂志, 2020, 58(6): 509–511.
[11] 梁伟玲, 刘陈菁, 吴倩, 等. 新发基因突变致腺苷脱氨酶缺乏症一例[J]. 中华儿科杂志, 2018, 56(8): 636–637.
[12] 王新宁, 周志轩, 李胜男, 等. 腺苷脱氨酶2缺乏症一例[J]. 中华风湿病学杂志, 2019, 23(7): 476–478.
[13] 柏翠, 郭兴青, 高婷婷, 等. 儿童腺苷脱氨酶2缺乏症2例并文献复习[J]. 中华实用儿科临床杂志, 2020, 35(21): 1674–1677.
[14] SAHIN S, ADROVIC A, KASAPCOPUR O. A monogenic autoinflammatory disease with fatal vasculitis: Deficiency of adenosine deaminase 2[J]. Curr Opin Rheumatol, 2020, 32(1): 3–14.
[15] GIBSON K M, MORISHITA K A, DANCEY P, et al. Identification of novel adenosine deaminase 2 gene variants and varied clinical phenotype in pediatric vasculitis[J]. Arthritis Rheumatol, 2019, 71(10): 1747–1755.
[16] CAORSI R, PENCO F, SCHENA F, et al. Monogenic polyarteritis: The lesson of ADA2 deficiency[J]. Pediatr Rheumatol Online J, 2016, 14(1): 51.
[17] WOOLLARD K J, GEISSMANN F. Monocytes in atherosclerosis: Subsets and functions[J]. Nat Rev Cardiol, 2010, 7(2): 77–86.
[18] MEYTS I, AKSENTIJEVICH I. Deficiency of adenosine deaminase 2 (DADA2): Updates on the phenotype, genetics, pathogenesis, and treatment[J]. J Clin Immunol, 2018, 38(5): 569–578.
[19] TROTTA L, MARTELIUS T, SIITONEN T, et al. ADA2 deficiency: Clonal lymphoproliferation in a subset of patients[J]. J Allergy Clin Immunol, 2018, 141(4): 1534–1537.
[20] BELOT A, WASSMER E, TWILT M, et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood[J]. Pediatr Rheumatol Online J, 2014, 12: 44.
[21] GARG N, KASAPCOPUR O, FOSTER J N, et al. Novel adenosine deaminase 2 mutations in a child with a fatal vasculopathy[J]. Eur J Pediatr, 2014, 173(6): 827–830.
[22] POSWAR F O, DA F R, DE ALBUQUERQUE L C, et al. Adenosine deaminase 2 deficiency presenting as spastic paraplegia and systemic vasculitis[J]. J Neurol, 2016, 263(4): 818–820.
[23]BARRON K S, AKSENTIJEVICH I, DEUITCH N T, et al. The spectrum of the deficiency of adenosine deaminase 2: An observational analysis of a 60 patient cohort[J]. Front Immunol, 2021, 12: 811473.
[24] CHUNN L M, BISSONNETTE J, HEINRICH S V, et al. Estimation of ENPP1 deficiency genetic prevalence using a comprehensive literature review and population databases[J]. Orphanet J Rare Dis, 2022, 17(1): 421.
Screening for carriers of pathogenic gene for deficiency of adenosine deaminase 2 in China
YAN Lulu, LOU Biying, XU Wenrong, LI Haibo
1.Laboratory of Birth Defects Prevention and Control, Ningbo Women & Children’s Hospital, Ningbo 315000, Zhejiang, China; 2.Medical School, Ningbo University, Ningbo 315000, Zhejiang, China; 3.Chigene (Beijing) Translational Medical Research Center Co. Ltd., Beijing, 100023
To analyze frequency of pathogenic gene for deficiency of adenosine deaminase 2 (DADA2) in China, so as to provide basis for effective prevention and intervention of DADA2.The original data of 111 000 subjects of whole exome sequencing performed in Chigene (Beijing) Translational Medical Research Center Co. Ltd. from January 2015 to January 2021 were retrospectively analyzed. The types of variants and allele frequency ofgene were analyzed.29 heterozygous variants ofgene were found. There were 6variants unreported previously, including c.730G>T (p.E244X), c.1049_c.1061delAGCTGCCTTACTT(p.K350Tfs*14), c.940A>T(p.K314X), c.1239+2(IVS8)T>C, c.1240-1(IVS8)G>A, c.1087C>T(p.Q363X), and the cumulative allele frequency ofwas 0.58%. The highest allele frequency of p.F355L was 0.5%, followed by p.Y453C (0.014%) and p.P193L (0.013%). Other 26 variant sites were less than 0.01%, indicating rare variant.It enriched the variational spectrum ofgene by the screening. The carrier frequency ofgene in China population was about 1 in 171 individuals, corresponding to an expected DADA2 disease prevalence of 1 in 116 964 individuals.
Deficiency of adenosine deaminase 2;gene; Allele frequency; Prevalence
R593.2
A
10.3969/j.issn.1673-9701.2023.25.005
浙江省医药卫生科技计划项目(2020KY890);宁波市科技计划项目(2022S035);宁波市医疗卫生高端团队重大攻坚项目(2022020405)
李海波,电子信箱:lihaibo-775@163.com
(2023–03–19)
(2023–08–19)



