吴 茗,刘丹如,俞晔珩,李 健,朱芬华
复旦大学附属儿科医院临床医学检验中心,上海 201102
先天性无肛属于先天性肛门直肠畸形(ARM)的一种,大约一半的ARM患者会合并其他器官系统的异常[1-4],但同时合并Kabuki综合征(KS)和生长激素(GH)缺乏导致身材矮小症的患儿却十分罕见。本院收治1例ARM合并KS伴身材矮小的儿童病例,现总结其发病特点、诊疗思路等,并进行文献复习,以期提高广大同行对该类疾病的认识,为临床诊疗提供参考依据。
1 临床资料
患儿,男,5岁10个月,2022年8月10日因发现身材矮小5年余到本院就诊。患儿家长自述出生时有宫内窘迫史、肾盂分离和卵圆孔未闭,成长后自愈。出生后诊断为先天性直肠缺如、闭锁和狭窄(无肛)伴KS。近5年来发现患儿身高低于同龄儿童,生长速度慢,遂就诊于本院。拟“身材矮小症”收入院。就诊时查体:患儿特殊面容,眼裂宽,低鼻梁,方颅,高鄂弓,肛门畸形术后改变,身高101 cm,体重16 kg,活动可,智力落后,听力未通过,戴助听器。能说句子,不流畅,运动、语言发育稍落后。否认喂养困难史,否认父母近亲结婚,无遗传性疾病史,无矮小家族史(男<160 cm,女<150 cm)。使用Illumina HiSeq平台进行高通量测序,与人类参考基因组序列对比,结果提示KMT2D基因新发杂合变异。见表1。血、尿、大便常规,肝、肾功能,血脂,电解质,肿瘤标志物,甲状腺功能检查均未见异常。胰岛素样生长因子1(IGF-1):44.60 μg/L,IGF结合蛋白3(IGF-BP3):3.30 μg/mL,GH:2.24 μU/mL。超声检查提示骨龄约2.5岁。磁共振检查提示Rathke囊肿,附见胼胝体膝部小囊性灶,双侧侧脑室局部稍扩张,胼胝体膝部可疑软化灶,双侧侧脑室扩张。患儿出生后发现肛门闭锁伴面容特殊,基因检测提示KMT2D基因新发杂合变异,诊断为KS 1型。患儿今年5岁10月,身高101 cm,低于同龄同性别同地区健康儿童身高的-2SD参考值,身材矮小、匀称,生长速度减慢,骨龄明显落后(落后2岁以上),GH激发试验提示GH缺乏,伴IGF-1减少,诊断为矮小症。患儿入院后完善各项辅助检查,空腹行可乐定激发试验,留取即刻时血液标本送检GH,予可乐定口服后分别留取30、60、90、120 min时血标本送检GH,实验有效,过程顺利,评估患儿GH恢复至正常水平后出院。见图1。
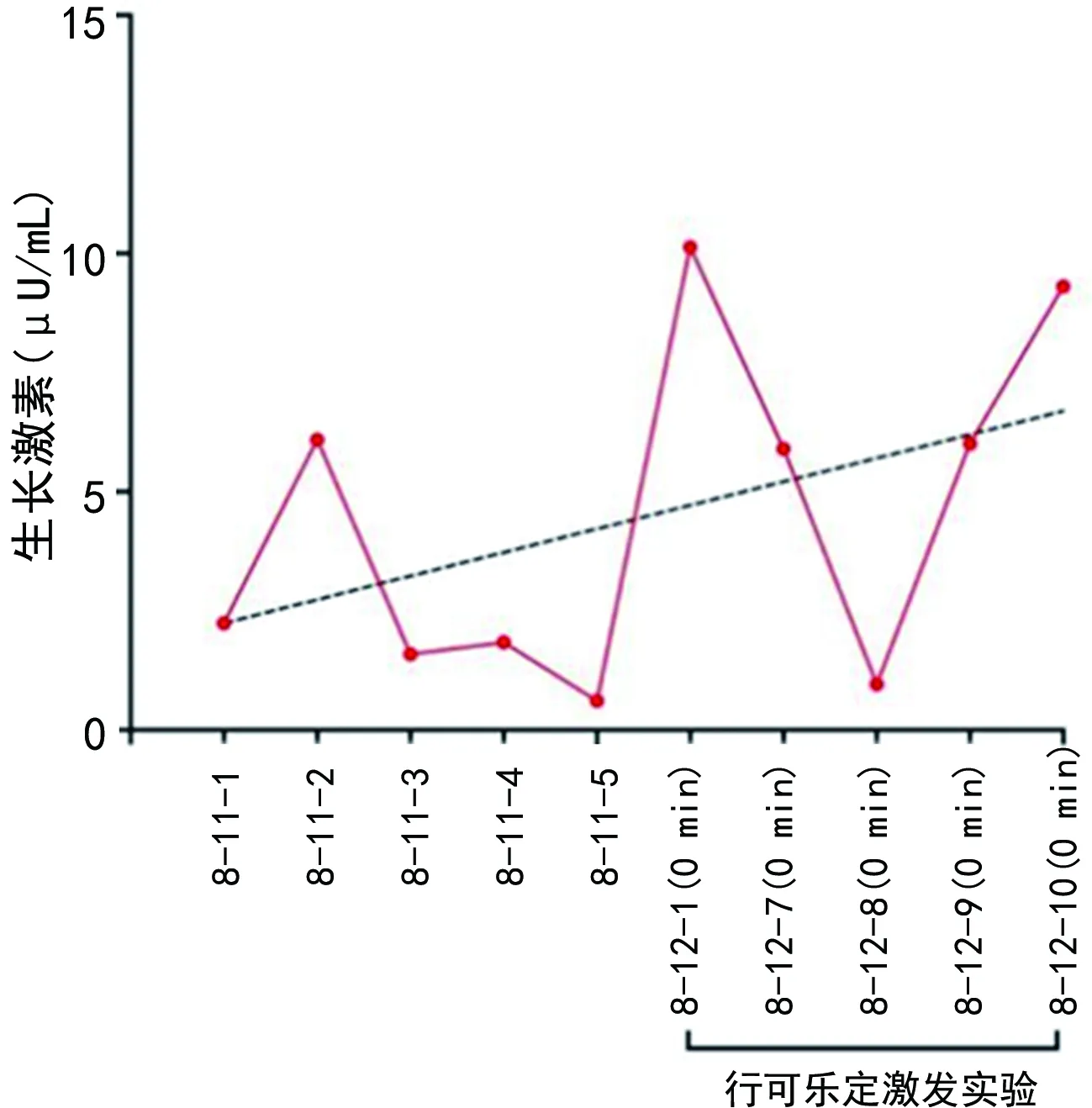
图1 住院期间血清GH检测结果变化趋势
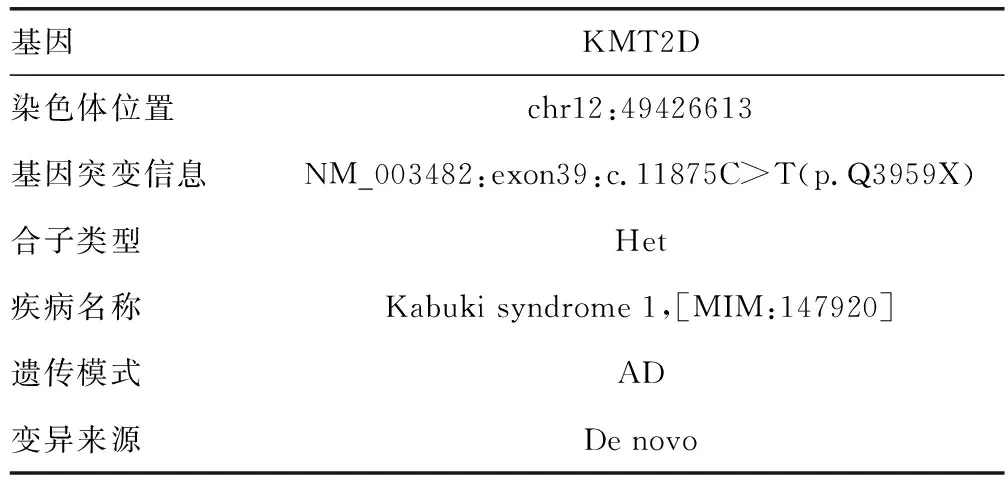
表1 基因检测结果
2 讨 论
大约每5 000个新生儿中会出现1例ARM,且男性多于女性[3],尽管大部分新生儿均会经过产后常规评估,但1/5的新生儿可能出现延迟诊断[2,4]。ARM常合并泌尿生殖系统和肌肉骨骼系统异常[1]。进一步对本例患儿进行基因检测进行排查。结果显示,患儿KMT2D基因新发杂合变异,提示KS 1型。KMT2D或KDM6A基因突变是导致KS发生的最主要原因,KS患儿中KMT2D基因突变率高达75%,KDM6A基因的突变占5%的病例(X连锁显性遗传),而约20%病例的病因仍然未知[5-6]。本例患儿经治疗出院后又因身材矮小5年余入院就诊,完善各项检查确诊为矮小症。有研究证实,50%~70%的KS病例出生后生长迟缓,有少数病例观察到产前生长迟缓,绝大多数病例观察到产后生长迟缓,且与种族无关[7]。此外,有学者指出,KMT2D基因突变的患者身材矮小发生率明显高于无KMT2D变异体的患者[8]。
以“Kabuki综合征、KMT2D和矮小”或“Kabuki Syndrome and Short Stature”为关键词检索中国知网、PubMed等数据库,检索时间为2012年10月1日至2022年10月1日,排除基础研究、综述、指南单表型研究。检索到11篇国人发表的文献[9-19],共24例中国KS同时伴有身材矮小(<-2SD)的患儿,纳入包括本例患儿共25例,男∶女=2.1∶1。应用人类表型本体论(HPO)对25例患儿进行临床表型分型,其中指尖垫(HPO:0001212)16例(64%),反复感染(HPO:0002205)15例(60%),外耳畸形(HPO:0040111)15例(60%),听力障碍(HPO:0000365)9例(36%),心脏异常(HPO:0002564)9例(36%),脊柱侧凸(HPO:0002650)8例(32%),喂食困难(HPO:0011968)8例 (32%),腭裂(HPO:0000175)5例(20%),肾脏异常(HPO:0000834)3例(12%),斜视(HPO:0000486)2例(8%),肛门闭锁(HPO:0002023)2例 (8%),GH缺乏(HPO:0000824)2例(8%),腺样体肥大(HPO:0040261)1例(4%),低血糖(HPO:0001943)例1(4%)。文献复习及本例患儿临床表征与上述研究结果较为一致,但大部分KS患儿的GH正常,GH缺乏合并各种出生缺陷极为罕见[20]。
本例患儿入院后完善辅助检查,进行精氨酸、可乐定激发试验,出院后暂予皮下注射长效生长激素(每周1次)或GH水剂(每晚1次),每3个月定期随访内分泌门诊,进行健康指导。SCHOTT等[21]发现,经过1年的重组生长激素(rh-GH)治疗后KS患者身高有了明显的线性增长,较早接受rh-GH治疗的儿童身高增加幅度更大,KMT2D组与KDM6A组无显着差异。但长期应用rh-GH治疗是否能继续改善身高、是否可普遍用于矮小且GH缺乏的KS患儿尚需深入研究,尤其是对合并多项出生缺陷基础疾病的患儿。
综上所述,本例患儿患有先天性ARM合并KS,同时患有卵圆孔未闭合肾盂分离等多种出生缺陷,生长后又因GH缺乏而矮小,且听力未通过,极为罕见。在临床诊疗中发现ARM或KS时一定要引起高度重视,务必考虑合并其他器官畸形可能,必要时进行详细检查,尤其是需要进行基因检测,对KS患儿应及时检测其各项代谢和生长指标,并及时进行干预治疗。此外对ARM患儿的手术选择应根据不同疾病特点制订个体化治疗方案。对KS合并矮小症患儿予以GH治疗及生活方式干预,应随时关注其内分泌代谢异常,通过长期随访内分泌门诊进行激素水平评估及正确的生活指导,及时发现潜在的风险并阻止其发生,以改善预后,有助于患儿获得良好的远期生活质量。



