王雨宁 郭晔堃 钟静芬
摘 要 已有甾体类醛固酮受体拮抗剂即螺内酯和依普利酮上市,但是两者均存在一定不足。非甾体的小分子化合物作为新一代的醛固酮受体拮抗剂,finerenone、esaxerenone等都已进入临床阶段,特别是finerenone,基础研究和临床研究显示出较优的安全性和有效性,在降低高钾血症和肾功能损伤方面具有独特的优势。此外,从醛固酮生物合成途径入手,靶标醛固酮生物合成过程中的关键酶即醛固酮合酶(CYP11B2),开发选择性的CYP11B2抑制剂也是当前研究的热点。
关键词 醛固酮 醛固酮受体 醛固酮合酶 抑制剂
中图分类号:R962 文献标志码:A 文章编号:1006-1533(2019)01-0056-06
Research progress in aldosterone receptor antagonists and aldosterone synthase inhibitors
WANG Yuning*, GUO Yekun**, ZHONG Jingfen
(China State Institute of Pharmaceutical Industry, Shanghai 201203, China)
ABSTRACT Although aldosterone receptor antagonists, such as spironolactone and eplerenone, are available, there still exist some disadvantages. Non-steroidal small molecules have been taken as a novel generation for aldosterone receptor antagonists. Finerenone and esaxerenone are all in clinical trial stage at present, especially for finerenone, its excellent safety and effectiveness have been verified by the basic and clinical studies, which has a unique advantage in lower hyperkalemia and renal injury. In addition, it was found that aldosterone synthase (CYP11B2) is the key one for aldosterone biosynthesis from the perspective of the aldosterone biosynthesis pathway. Nowadays, the development of selective CYP11B2 inhibitors is also a research hotspot.
KEy WORDS aldosterone; aldosterone receptor; aldosterone synthase; inhibitor
1 醛固酮的生理病理作用
人类肾上腺皮质球状带区域分泌有盐皮质激素,醛固酮是一种盐皮质激素,能促进肾远曲小管对钠离子、氯离子的重吸收和增加钾离子、氢离子的排出,具有明显的潴钠排钾的作用。醛固酮可以通过提升肾远曲小管对钠离子的重吸收来调控血压。在正常生理条件下,醛固酮的分泌受肾素-血管紧张素-醛固酮系统(RAAS)调节,另外心血管独立存在的醛固酮形成系统也可以使醛固酮以自分泌和旁分泌的形式在局部发挥作用。
醛固酮水平过高会造成心肌及血管间质纤维化,导致心室重构,血管壁增厚,大动脉顺应性降低,心脏功能恶化,使组织传导不均一,引发心律失常[1]。醛固酮还可以阻断心肌细胞对儿茶酚胺的摄取,使细胞外儿茶酚胺增加,加重心肌缺血[2]。有研究表明醛固酮含量过高时会诱发白细胞浸润并会造成冠状动脉损伤以及心肌缺血性坏死。有研究显示,心肌梗死的表达和进程会促进炎症因子骨调素(OPN)、单核细胞趋化因子(MCP-1)和环氧合酶-2(COX-2)对醛固酮的应答从而引起血管炎症[3]。
2 醛固酮受体拮抗剂的应用
竞争性阻断醛固酮与受体结合,即拮抗醛固酮对Na+的重吸收和K+的排出,使Na+、水排出增多,尿量增加,K+排出减少,起到潴钾利尿的作用。目前,已有临床试验证实血管紧张素转化酶抑制剂(ACEIs)或血管紧张素受体阻断剂(ARBs)与醛固酮受体阻断剂联合治疗降压效果更明显,特别是针对顽固性高血压的治疗。此外,长期使用ACEIs或ARBs之后,会出现醛固酮水平升高甚至超过治疗前水平的情况,此时醛固酮受体拮抗剂可以拮抗醛固酮水平过高而对心脏结构和功能的不良作用,因此目前更提倡ACEIs或ARBs联合醛固酮受体拮抗剂治疗高血压与心力衰竭。
3 醛固酮合酶(CYP11B2)抑制剂的治疗机制
病理状态下,醛固酮水平意外升高,加之长期使用ACEIs或ARBs会造成血管紧张素Ⅰ含量反馈性地增加,从而其通过旁路合成血管紧张素Ⅱ,进而刺激醛固酮的分泌,我们设想可以从源头上减少醛固酮的合成从而减弱醛固酮效应。
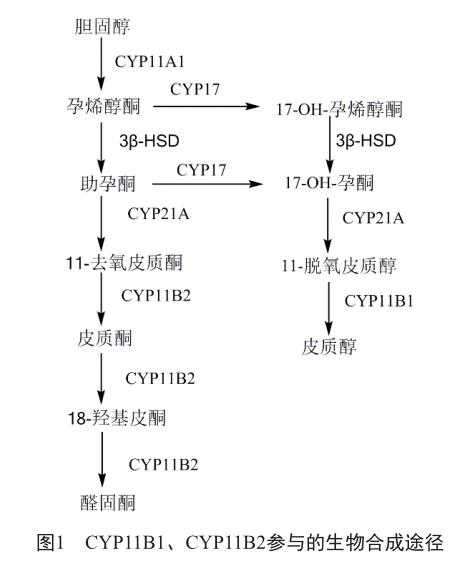
CYP11B2是一种线粒体细胞色素,催化醛固酮生物合成途径的最后三步,其还有另一种同工酶即11β-羟化酶(CYP11B1),两者的同源序列高达93%[4],且都分布在肾上腺皮质区。CYP11B1(图1)、CYP11B2(图1)分别是参与皮质醇和醛固酮的生物合成最后环节中的关键酶,抑制CYP11B1可以作为一种治疗与皮质醇水平意外升高相关疾病的途径,例如库欣综合征、代谢疾病和伤口延迟愈合[5]。CYP11B2是醛固酮生物合成过程中的关键酶,抑制CYP11B2是治疗充血性心力衰竭、心肌纤维化和特定形式高血压的潜在方案[6]。
4 醛固酮受体拮抗剂
4.1 甾体类
4.1.1 螺内酯
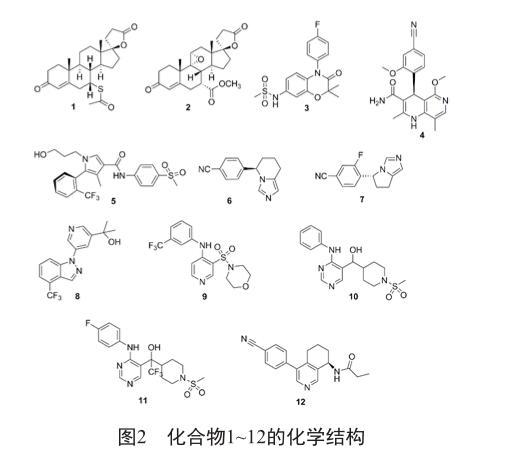
螺内酯(1)(图2)于1960年上市批准作为利尿剂用于水肿、原发性醛固酮症和原发性高血压患者的治疗。随后又有临床试验数据显示螺内酯用于严重心力衰竭患者可以显着降低总死亡率、心脏病死亡率、心力衰竭恶化致死率、猝死率以及心力衰竭恶化住院率[7]。但是由于其会在一定程度上结合雄激素受体和黄体酮受体从而分别导致男子女性型乳房和女子月经不调,另外其对肾损伤病人会引发高钾血症的潜在生命危险。
4.1.2 依普利酮
与螺内酯不同的是,依普利酮(2)(图2)在9,11位以环氧基取代,7位以甲氧甲酰基取代,是一类全新的醛固酮受体拮抗剂,这种环氧结构的修饰使得这个化合物呈现凹面的立体结构,因而它能选择性地阻滞盐皮质激素受体而对糖皮质激素受体和孕酮或雄激素受体无明显影响[8]。该药物半衰期较长,每日口服一次就可有效地控制高血压,减轻心、脑和肾等靶器官的损害,改善2型糖尿病患者微量蛋白尿,而且其副作用发生率除了血钾水平升高外与安慰剂相似,耐受良好。另外,依普利酮还可以提高左心室功能紊乱(射血分数≤40%)患者的生存质量,临床试验数据显示对急性心肌梗死后心力衰竭的治疗,和标准治疗药物联合使用可以使治疗组总死亡率降低15%,证明该药物可以用于急性心肌梗死后心力衰竭的治疗[9]。
4.2 非甾体类
对于心力衰竭的治疗,在1980年之后逐渐由利尿剂和强心苷类转化至神经体液调节药物,例如ACEIs、b受体阻断剂和醛固酮受体拮抗剂[10]。螺内酯是第一个上市的醛固酮受体拮抗剂,但是服用螺内酯特别是结合ACEI使用有一个潜在危害就是会诱发具有生命隐患的高钾血症[11],肾功能损伤或者存在高钾血症的患者不适合使用螺内酯和依普利酮。最受关注的是螺内酯对盐皮质激素受体活性较高但是选择性太低,而依普利酮选择性较高但是对盐皮质激素受体的活性太低,我们寄希望于同时具有高选择性和高活性的并且安全性更高的醛固酮受体拮抗剂。我们在此介绍已经进入临床阶段的三个在研化合物,apararenone(3)、finerenone(4)、esaxerenone(5)(图2)。
4.2.1 apararenone (MT-3995)
aparareone是由日本田边三菱制药开发的非甾体类选择性拮抗盐皮质激素的药物,目前其正在东欧和日本进行用于治疗糖尿病肾病的二期临床试验以及在日本进行用于治疗非酒精性脂肪肝炎的二期临床试验[12],前期开发用于高血压适应证的治疗已经终止。
4.2.2 finerenone(BAY 94-8862)
4.2.2.1 临床前研究
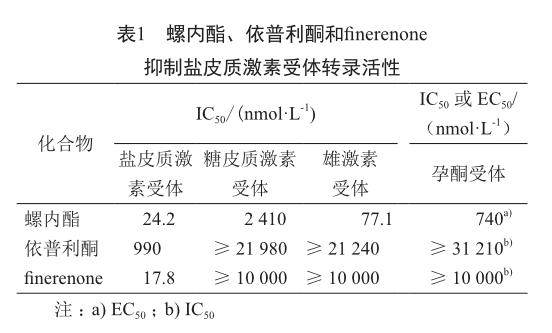
由于Ca2+通道阻断剂二氢吡啶类化合物在体外具有抗盐皮质激素受体的良好活性[13],对其结构修饰可得到类似物二氢萘啶化合物finerenone。在以细胞为基础的甾体激素受体转录活性试验中,主要比较螺内酯、依普利酮和finerenone的活性和选择性(表1),结果表明finerenone拮抗盐皮质激素受体活性要强于螺内酯和依普利酮,选择性要高于螺内酯[14]。
在去氧皮质酮醋酸盐大鼠高血压模型中,finrenone在10 mg/kg的剂量下表现出降低收缩压的活性,并且收缩压要低于在100 mg/kg的剂量下的依普利酮组。在高压诱导的心力衰竭模型中,采用finerenone治疗抑制心肌肥厚的活性大于依普利酮[15]。
4.2.2.2 临床研究
finerenone与螺内酯和依普利酮不同之处在于它有均衡的组织分布,大鼠模型中结果显示其在心脏、肾分布几乎相等[16]。在慢性心力衰竭和轻度或中度慢性肾病的患者中开展的用于评价finerenone的安全性和耐受性临床试验中,finerenone各剂量组血液中钾离子水平升高为0.04~0.30 mmol/L,而螺内酯为0.45 mmol/L,并且finerenone剂量组中高钾血症和肾功能损伤发生率更低。finerenone降低B型利钠肽、脑钠肽和蛋白尿水平与螺内酯相当[17]。
在接受过ACE抑制剂或者血管紧张素受体抑制剂的糖尿病肾病患者中进行的随机临床试验,用于评价不同剂量下finerenone降低患者蛋白尿的安全性和有效性,结果显示,finerenone呈浓度相关性降低白蛋白-肌酸酐比率,如果以安慰剂组结果为基线1,finerenone在剂量为7.5,10,15和 20 mg/d下尿白蛋白与肌酐比值(UACR)水平下降分别为0.79,0.76,0.67和0.62,并且在安慰剂和10 mg/d的剂量组中没有发生由于高钾血症引起的终止试验案例[18]。
在恶化的慢性心力衰竭和糖尿病或肾病患者中开展的对比finerenone和依普利酮疗效的临床试验中,依普利酮组中降低脑钠肽水平超过30%的患者数占37.2%,finerenone剂量为2.5~5、5~10、7.5~15、10~20和15~20 mg组结果分别为30.9%、32.5%、37.3%、38.8%和34.2%。另外,finerenone剂量为10~20 mg的组别中,各项原因造成的死亡、心血管住院率以及紧急情况住院率最低[19]。此次临床试验显示出10~20 mg的剂量下,无论是安全性还是有效性都是对患者最有保障的,这为以后展开更大规模的临床试验提供数据支撑。
在评价finerenone对糖尿病肾病患者隐匿性高血压和夜间高压疗效的随机试验中,10 mg和20 mg剂量组降低隐匿性高压为正常值的患者比例分别为60%和66.7%,而安慰剂组结果是40%,两个剂量组降低夜间高压为正常值的患者比例为37.5%,安慰剂组结果为0%。另外,夜间高压患者在服用10 mg、20 mg finerenone后夜间高压下降值分别为9.5 mmHg和12.0 mmHg,安慰剂组夜间高压减少值只有2.9 mmHg[20]。
4.2.3 esaxerenone(CS-3150)
4.2.3.1 临床前研究
在报告基因试验中,esaxerenone抑制人类盐皮质激素受体转录活性的IC50值是3.7 nmol/L,螺内酯和依普利酮的IC50值分别是66 nmol/L和970 nmol/L,esaxerenone的活性优于螺内酯和依普利酮,而且即便在5 mmol/L的浓度下,esaxerenone也并没有呈现出抑制糖皮质激素受体、雄激素受体和孕酮受体的活性[21](表2)。
在放射性配体结合试验中,esaxerenone呈浓度相关性抑制放射性配体3H-醛固酮与盐皮质激素受体的结合,IC50值为9.4 nmol/L,而螺内酯和依普利酮分别是36 nmol/L和713 nmol/L,esaxerenone与盐皮质激素受体的亲和性是螺内酯的4倍,是依普利酮的76倍。另外,仅在esaxerenone浓度为10 mmol/L时,糖皮质激素受体、雄激素受体和孕酮受体才表现出轻微抑制性,这项研究表明esaxerenone对盐皮质激素受体的亲和性要比其它三种受体高出1 000倍[21](表3)。
在去氧皮质酮醋酸盐小鼠高血压模型中,esaxerenone在3 mg/kg的剂量下就可以显着降低心脏收缩压,但是螺内酯和依普利酮在30 mg/kg的剂量下都无法达到此类效应,esaxerenone抗高血压的活性优于螺内酯和依普利酮[21]。
esaxerenone可以抑制高盐饮食诱发的蛋白尿和肾肥大[22]。对肾脏的组织病理学检查表明其可以显着减缓肾小球硬化症、肾小管损伤和肾小管间质纤维化。另外有研究表明esaxerenone可以抑制左心室肥大以及降低脑血浆利钠肽水平[23]。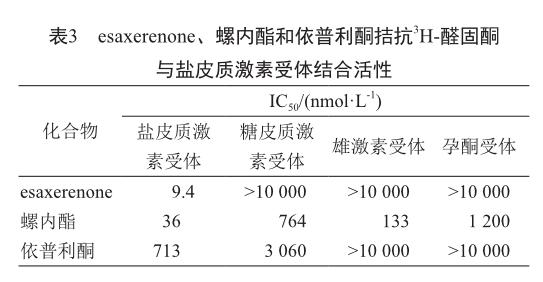
4.2.3.2 临床研究
2015年2月,第一三共宣布其要开展两项二期临床试验:在日本用于2型糖尿病患者和微蛋白尿患者的关于esaxerenone的剂量范围研究和日本高血压患者身上服用esaxerenone的有效性和安全性研究。2016年9月,另外一项关于评价esaxerenone用于治疗原发性高血压的重要三期临床试验宣布正在进行中,其主要目的就是为了说明其抗高血压的效应不亚于依普利酮。
5 醛固酮合酶抑制剂
螺内酯和依普利酮的各项临床试验显示,醛固酮受体拮抗剂可以降低充血性心力衰竭和急性后期心肌梗死患者的致死率[24]。但研究也显示,药物对醛固酮受体的拮抗作用会使得血浆中醛固酮浓度上升以及醛固酮合成增加。另有研究表明盐皮质激素受体拮抗剂无法拮抗独立于受体的醛固酮的非基因组活性[25]。此外,盐皮质激素受体拮抗剂存在另一缺陷是高钾血症,需对患者血钾水平进行监测。因此,直接抑制醛固酮生成的新化合物,逐渐成为这一领域研究的热点。
5.1 先导化合物
非甾体芳香化酶抑制剂法曲唑的立体异构体FAD-286(6)(图2)可以抑制人类重组CYP11B1(IC50=9.9 nmol/L)和CYP11B2(IC50=1.6 nmol/L)。在高表达人类肾素和血管紧张素原基因的大鼠(dTGR)模型中,FAD-286可以抑制CYP11B2,减少循环系统和心脏醛固酮水平并改善内皮细胞功能,减少心脏和肾靶器官损害[26]。
在FAD-286的基础上进行结构改造又得到LCI-699(7)(图2)。在14例原发性醛固酮增多症患者中开展的概念验证研究中,LCI-699剂量为0.5 mg和1.0 mg时可以显着降低患者仰卧位血浆醛固酮浓度[27]。在dTGR中,LCI-699呈浓度相关性阻断醛固酮的合成,阻止与血压变化物无关的心脏和肾功能恶化从而延长寿命,依普利酮相也有类似作用但相对较弱[27]。
在0.5 mg剂量下,LCI-699可以选择性地抑制醛固酮的合成,且不影响皮质醇的合成,但当剂量大于1 mg时,对CYP11B2的选择性就会明显下降[28]。
5.2 其它杂环衍生物
5.2.1 苯并吡唑衍生物
Whitehead等[29]认识到CYP11B2抑制剂除了应具有良好的抑制活性外对CYP11B1的选择性亦很重要。研究小组以吡啶环取代原有的苯环,设计并合成了一系列吡啶并咪唑化合物和苯并吡唑化合物。Hoyt等[30]发现化合物8(图2)具有抑制CYP11B2的良好活性,其对CYP11B2和CYP11B1的IC50值分别是2.2 nmol/L和98 nmol/L,且相较于其它肝微粒体酶系选择性较高。
5.2.2 嘧啶化合物
Meguro等[31]在化合物库中成功筛选出化合物9(图2),其具有对CYP11B2的高抑制活性,但是由于其对药物相关代谢酶的抑制性以及较差的代谢稳定性,对其进行结构改造,用羟甲基替代原来的磺酰基改善代谢稳定性,用嘧啶环替代吡啶环减轻对相关药物代谢酶CYP3A4的抑制性,得到化合物10(图2)和化合物11(图2),后者在体外试验中在纳摩尔级浓度便可以抑制人类和啮齿类动物CYP11B2,在呋塞米处理的猕猴模型中具有降低醛固酮的效应。
5.2.3 四氢异喹啉化合物
Martin等[32]发现新型四氢异喹啉化合物12(图2)在具有良好的抑制人类和猕猴的醛固酮合酶的活性,并且在猕猴试验中,CYP11B2/CYP11B1的选择系数达到160,它可以呈浓度相关性显着降低血浆中醛固酮水平,在ZSF1慢性肾疾病大鼠模型(由糖尿病肥胖大鼠和自发性高血压心衰大鼠杂交产生)中,它还可以保护肾脏结构和功能。
6 讨论
螺内酯和依普利酮分别被认为是第一代和第二代醛固酮受体拮抗剂,但是两者在临床上并不具有独特的优势,finerenone在相关基础研究和临床研究中显示出其高选择性和对醛固酮受体的高亲和力,并且展现出了较好的安全性,可用于严重肾功能损伤的患者,可减少高钾血症和肾功能损伤的危害。esaxerenone在相关基础研究上显示出其对醛固酮受体的高选择性和亲和力,其在临床阶段的治疗效果还需要更多数据的支撑。
与醛固酮受体拮抗剂相比,醛固酮合酶抑制剂可以抑制ACEIs或者ARBs导致的醛固酮水平升高而引发的效应,也可以抑制靶器官中独立于醛固酮受体或者依赖于受体的醛固酮效应。尽管醛固酮合酶抑制剂可以作为醛固酮受体拮抗剂之外的选择,但是其最大的挑战可能在于其要证实在高血压、心力衰竭或者肾脏疾病方面具有更好的疗效和耐受性。
参考文献
[1] 王正, 沈娟, 宋庆桥. 慢性心力衰竭合并心律失常发病机制研究进展[J]. 中西医结合心脑血管病杂志, 2012, 10(2): 216-218.
[2] 魏顺利, 谷新顺, 姜春素, 等. 国产依普利酮对家兔急性心肌梗死后心功能的影响[J]. 河北医药, 2010, 32(22): 3109-3111.
[3] Rocha R, Rudolph AE, Frierdich GE, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart[J]. Am J Physiol-Heart C, 2002, 283(5): H1802-H1810.
[4] Belkina NV, Lisurek M, Ivanov AS, et al. Modelling of three-dimensional structures of cytochromes P450 11B1 and 11B2[J]. J Inorg Biochem, 2001, 87(4): 197-207.
[5] Zhu W, Chen Z, Li Q, et al. Inhibitors of 11β-hydroxylase(CYP11B1) for treating diseases related to excess cortisol[J]. Curr Med Chem, 2016, 23(6): 623-633.
[6] Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system[J]. Nat Rev Nephrol, 2010, 6(5): 261-273.
[7] Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure[J]. New Engl J Med, 1999, 341(10): 709-717.
[8] Grob J, Boillaz M, Schmidlin J, et al. Steroidal, aldosterone antagonists: increased selectivity of 9α, 11-epoxy deriva-tives[J]. Helv Chim Acta, 1997, 80(2): 566-585.
[9] Zannad F, McMurray JJV, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms[J]. New Engl J Med, 2011, 364(1): 11-21.
[10] Hunt SA, American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure)[J]. J Am Coll Cardiol, 2005, 46(6): e1-e82.
[11] Berry C, McMurray JJV. Serious adverse events experienced by patients with chronic heart failure taking spironolactone[J/ OL]. Heart, 2001, 85(4): e8. doi: 10.1136/heart.85.4.e8.
[12] Kolkhof P, B?rfacker L. 30 years of the mineralocorticoid receptor: mineralocorticoid receptor antagonists: 60 years of research and development[J/OL]. J Endocrinol, 2017, 234(1): T125-T140. doi: 10.1530/JOE-16-0600.
[13] Ergueden JK, Kolkhof P, Sandner P, et al. Fluorenone 1,4,-dihydropyridin derivatives: WO 2005087740A1[P]. 2005-09-22.
[14] Pitt B, Filippatos G, Gheorghiade M, et al. Rationale and design of ARTS: a randomized, double‐blind study of BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease[J]. Eur J Heart Fail, 2012, 14(6): 668-675.
[15] Grune J, Benz V, Brix S, et al. Steroidal and nonsteroidal mineralocorticoid receptor antagonists cause differential cardiac gene expression in pressure overload-induced cardiac hypertrophy[J]. J Cardiovasc Pharm, 2016, 67(5): 402-411.
[16] B?rfacker L, Kuhl A, Hillisch A, et al. Discovery of BAY 94-8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases[J]. Chem Med Chem, 2012, 7(8): 1385-1403.
[17] Pitt B, Kober L, Ponikowski P, et al. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, doubleblind trial[J]. Eur Heart J, 2013, 34(31): 2453-2463.
[18] Bakris GL, Agarwal R, Chan JC, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial[J]. JAMA, 2015, 314(9): 884-894.
[19] Filippatos G, Anker S D, B?hm M, et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease[J]. Eur Heart J, 2016, 37(27): 2105-2114.
[20] Ruilope LM, Nowack C, Bakris GL. Masked and nocturnal hypertension in the ARTS-DN ABPM sub-study with finerenone[J]. J Am Soc Hypertens, 2016, 10(Suppl 1): e1-e9.
[21] Arai K, Homma T, Morikawa Y, et al. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist[J]. Eur J Pharmacol, 2015, 761: 226-234.
[22] Arai K, Tsuruoka H, Homma T. CS-3150, a novel nonsteroidal mineralocorticoid receptor antagonist, prevents hypertension and cardiorenal injury in Dahl salt-sensitive hypertensive rats[J]. Eur J Pharmacol, 2015, 769: 266-273.
[23] Arai K, Morikawa Y, Ubukata N, et al. CS-3150, a novel nonsteroidal mineralocorticoid receptor antagonist, shows preventive and therapeutic effects on renal injury in deoxycorticosterone acetate/salt-induced hypertensive rats[J]. J Pharmacol Exp Ther, 2016, 358(3): 548-557.
[24] Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure[J]. New Engl J Med, 1999, 341(10): 709-717.
[25] Wendler A, Albrecht C, Wehling M. Nongenomic actions of aldosterone and progesterone revisited[J]. Steroids, 2012, 77(10): 1002-1006.
[26] Fiebeler A, Nussberger J, Shagdarsuren E, et al. Aldosterone synthase inhibitor ameliorates angiotensin II–induced organ damage[J]. Circulation, 2005, 111(23): 3087-3094.
[27] Amar L, Azizi M, Menard J, et al. Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism[J]. Hypertension, 2010, 56(5): 831-838.
[28] Andersen K, Hartman D, Peppard T, et al. The effects of aldosterone synthase inhibition on aldosterone and cortisol in patients with hypertension: a phase II, randomized, doubleblind, placebo-controlled, multicenter study[J]. J Clin Hypertens, 2012, 14(9): 580-587.
[29] Whitehead BR, Lo MM, Ali A, et al. Imidazopyridyl compounds as aldosterone synthase inhibitors[J]. Bioorg Med Chem Lett, 2017, 27(2): 143-146.
[30] Hoyt SB, Taylor J, London C, et al. Discovery of indazole aldosterone synthase (CYP11B2) inhibitors as potential treatments for hypertension[J]. Bioorg Med Chem Lett, 2017, 27(11): 2384-2388.
[31] Meguro M, Miyauchi S, Kanao Y, et al. 4-Anilino-pyrimidine,novel aldosterone synthase (CYP11B2) inhibitors bearing pyrimidine structures[J]. Bioorg Med Chem Lett, 2017, 27(9): 1902-1906.
[32] Martin RE, Aebi JD, Hornsperger B, et al. Discovery of 4-Aryl-5,6,7,8-tetrahydroisoquinolines as potent, selective, and orally active aldosterone synthase (CYP11B2) inhibitors: in vivo evaluation in rodents and cynomolgus monkeys[J]. J Med Chem, 2015, 58(20): 8054-8065.



