

(2020-05-20发布2020-07-01实施)
前言
本文件按照GB/T 1.1-2020《标准化工作导则第1部分:标准化文件的结构和起草规则》的规定起草。
本文件是窄治疗指数药物质量评价及标准制订的技术性指导文件。
本文件由上海医药行业协会提出。
本文件由上海医药行业协会归口。
本文件起草单位:上海医药行业协会、上海药品审评核查中心、上海市第六人民医院、赛诺菲(杭州)制药有限公司。
本文件主要起草人:陈桂良、张景辰、陈莉莉、刘朋、陈志东、王子涛、阮克萍、林毅楠、黄一帆、荑征宇、肇晖、吴耀卫、朱蓓芬。
本文件首批执行单位:赛诺菲(杭州)制药有限公司、上海复旦复华药业有限公司、上海禾丰制药有限公司、上海上药第一生化药业有限公司、上海上药中西制药有限公司、上海上药信谊药厂有限公司、上海新亚药业闽行有限公司、默克雪兰诺有限公司。
本文件为首次发布。
引言
窄治疗指数药物是指当剂量或血药浓度发生小的差异时,可能导致严重治疗失败和/或药物不良反应(即可危厦生命或导致持久的或严重伤残)的药物。
目前,我国尚未建立完善的针对窄治疗指数药物全生命周期管理的制度和技术规范。鉴于此,上海医药行业协会提出制订此标准,以期加强对窄治疗指数药物的管理,保证人民用药安全。
由于窄治疗指数药物的特殊性,本文件4.3窄治疗指数药物的质量评价,重点对窄治疗指数药物人体生物等效性试验和稳定性研究两方面进行了规定。
本文件附录C窄治疗指数药物目录中收载品种的遴选,参照了国内外相关目录和文献资料,且为首批执行单位品种。
1范围
本文件适用于窄治疗指数药物质量评价及标准制订。
2规范性引用文件
下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
中华人民共和国药品管理法中华人民共和国第十三届全国人民代表大会常务委员会
药品生产质量管理规范中华人民共和国卫生部
中华人民共和国药典国家药典委员会
人用药品注册技术要求协调会议指导原则QIE Q6、QlO Q12(IntemationaICmmcilfor Hmmonization guideline)
美国药典(US Phmmacoepia)美国药典委员会
英国药典(British Pharmacoepia)英国药品委员会
3术语和定义
下列术语和定义适用于本文件。
3.1半数致死量(Median Lethal Dose,简称LD50)
能引起50%的实验动物出现死亡反应时的药物剂量。
3.2半数有效剂量(Median Effective Dose,简称ED50)
能引起50W0的实验动物出现阳性反应时的药物剂量。
3.3治疗指数(Therapeutic Index,简称TI)
药物的LD50/ED50的比值称为治疗指数,用于表示药物的安全性。
3.4窄治疗指数(Narrow Therapeutic Index,简称NTI)
T1小于2,或最小中毒血药浓度和最小有效血药浓度之比小于2。
3.5窄治疗指数药物(Narrow Therapeutic Index Drugs,简称NTIDs)
NTIDs是指当剂量或血药浓度发生小的差异时,可能导致严重治疗失败和/或药物不良反应(即可危及生命或导致持久的或严重伤残)的药物。一般情况是指符合T1小于2的药物。
3.6药物全生命周期(Life-Cycle Management)
药物研发、技术转移、商业生产、流通使用直至产品终止的生命周期。
4技术要求
4.1总则
4.1.1本技术的制定参考了有关窄治疗指数药物相关的法律、法规和技术要求,并以现有窄治疗指数药物的研发、生产、流通、使用的研究数据和临床监测数据为依据。
4.1.2基于NTIDs特性及临床使用的风险,本文件以质量管理为核心,从质量评价和质量标准的建立等方面进行了规定。
4.2窄治疗指数药物的质量管理
4.2.1应当对NTIDs的全生命周期进行严格的质量管理,确保药物的关键质量属性在全生命周期各个环节都能够得到有效的维护和传递。
4.2.2 NTIDs全生命周期质量管理的核心是建立质量保证体系,用于确保药物的安全性、有效性和质量可控性。
4.2.2.1质量保证体系的设计、建立和文件编制应当具有可执行性且清晰明确,以确保理解和执行的一致性。
4.2.2.2质量保证体系应当有效地运用全生命周期的知识管理和质量风险管理,为产品质量提供相关的科学和风险的决策方法。通过对质量保证体系的持续改进,降低药物质量的波动,推动产品质量不断提升。
注:知识管理是指从产品的开发到产品的商业生命周期,直至产品停产,都应该对产品和过程知识进行管理。例如,科学方法指导的开发活动为理解产品和开发过程提供了知识。知识管理是获取、分析、存储和传播与产品、制造过程和部件有关的信息的系统方法。知识来源包括但不限于先前的知识(公开文件或内部文件)、药物开发研究、技术转让活动、产品生命周期的过程验证研究、制造经验、创新、持续改进和变更管理活动。
4.2.2.3质量保证体系应重点关注对工艺性能、剂型剂量、药代动力学、生物等效性评价等质量评价核查,以及产品质量监测系统、纠正和预防措施系统、变更管理系统、工艺性能和产品质量的管理评审、药物警戒、临床合理应用、用药安全监测、药品信息化追溯等方面的管理。
4.2.2.4应当定期对质量体系运行的适宜性、充分性、有效性进行回顾,并做出评价,确保质量体系持续得到改进。
4.2.3 NTIDs的上市许可持有人,应建立良好质量评价体系和风险防控体系,确保在全生命周期中能够有效地进行监测、评价和控制。
4.2.3.1应当制定药物上市后风险管理计划,主动开展药物上市后研究,对药物的安全性、有效性和质量可控性进行进一步确证。
4.2.3.2应当加强对已上市药物的持续管理,主动收集、跟踪分析疑似药物不良反应信息,遇到特殊情况能够及时采取有效的措施确保药物质量和风险最小化。
4.2.4 NTIDs在研发过程中,应重点关注用药剂量与血药浓度检测相关资料(包括药剂、药理、毒理、生物等效性试验数据),须充分证明药物剂量、药物释放行为和含量控制范围的合理性。
4.2.5 NTIDs在生产过程中,应建立良好的质量保证体系,用于确保能够持续生产出严格符合质量目标的药品。
4.2.6 NTIDs在流通过程中,采购、保管、调配宜实行严格管理。应建立良好的质量保证体系,确保药物在流通过程中持续符合质量要求,全程可追溯。
4.2.7应当加强NTIDs的临床使用管理,以确保临床使用安全。
4.2.7.1 NTIDs在临床使用过程中,各医疗机构应当建立标准化的管理制度、有明确的药物警戒报告制度和药物安全监测实施方案,同时加强对相关责任人的教育培训。
4.2.7.2 NTIDs处方的医生,应按照药品说明书用药。特殊情况下的超说明书用药,必须要符合有关规定。谨慎选择与替换不同厂家的NTIDs。
4.2.7.3药师针对NTIDs为患者提供药学服务,应明确告知药物的用法用量、不良反应风险及所需开展的用药安全监测方案,加强处方的审核、调配等技术服务。
4.3窄治疗指数药物的质量评价
4.3.1窄治疗指数药物人体生物等效性试验
4.3.1.1 NTIDs人体生物等效性试验,一般包括试验前期设计、受试者选择、参比制剂选择、单次给药研究、稳态研究、餐后生物等效性研究、生物样品分析、用于评价生物等效性的药代动力学参数及生物等效性试验实施过程及数据统计分析等。
4.3.1.2 NTIDs生物等效性试验推荐考虑四向、全重复、交叉研究设计,以评估受试制剂与参比制剂的个体内变异情况(within-subject variability,WSV),并使用参比制剂标度平均生物等效性(reference-scaled average bioequivalence,RSABE)方法,对相关药代动力学参数进行统计比较。
4.3.1.3对比受试制剂和参比制剂的wsv情况,同时保留非标度的平均生物等效评价来确定各个药代动力学参数的生物等效,即生物等效性所有的研究既要通过参比制剂标定的生物等效限,又必须通过常规的非标定生物等效限。
4.3.1.4 RSABE方法下,应根据药物的WSV特性,适当缩窄90%置信区间范围,具体范围值可通过RSABE方法计算得出。
4.3.2窄治疗指数药物稳定性研究
4.3.2.1 NTIDs稳定性研究是质量控制研究的重要组成部分,应当按照《中华人民共和国药典》四部9001原料药物与制剂稳定性试验指导原则开展。
4.3.2.2稳定性试验通常包括影响因素试验、加速试验和长期试验。
4.3.2.2.1影响因素试验主要是了解其对光、湿、热、酸、碱、氧化等环境的敏感性,主要的降解途径及降解产物,并据此为进一步验证所用分析方法的专属性、确定加速试验的放置条件及选择合适的包装材料提供参考。
4.3.2.2.2加速试验是考察原料药或制剂在高于长期贮藏温度和湿度条件下的稳定性,为处方工艺设计、偏离实际贮藏条件其是否依旧能保持质量稳定提供依据,并根据试验结果确定是否需要进行中间条件下的稳定性试验及确定长期试验的放置条件。
4.3.2.2.3长期试验则是考察原料药或制剂在拟定贮藏条件下的稳定性,为确认包装、贮藏条件及有效期提供数据支持。
4.3.2.3 NTIDs稳定性研究试验条件的选择应当充分考虑其特性,确保涵盏生产、流通、使用过程中的实际存放条件。
4.3.2.4 NTIDs应当根据其具体的临床使用情况,对“临用现配”的制剂或“多剂量包装”开启后,有一定的使用期限的制剂,进行配伍稳定性试验或开启后使用的稳定性试验。
4.3.2.5对稳定性试验结果进行分析评估时,应当考虑NTIDs原料药和制剂关键质量属性的变化趋势和批次间的质量波动。
4.3.2.6如果稳定性数据显示原料药和制剂关键质量属性存在变化趋势且批次间有一定的质量波动,则应采用统计分析的方法对这些差异进行评估。应当重点关注数据变化趋势的合理性,数据发生异常变化的时间点及试验条件等。
4.3.2.7进行评估的定量指标应当考虑活性成分的含量、含量均匀度、降解产物的水平、溶出度或释放度及其他有关的质量属性。重点关注与参比制剂稳定性数据变化趋势之间的差异。必要时,还应关注质量守恒原则、稳定性试验数据波动和降解特性。
4.3.2.8根据稳定性试验数据分析的结果,持续完善NTIDs的处方、工艺及质量标准。
4.3.2.9对会随时间变化的定量指标进行统计分析,具体方法参照ICH QIE或国家发布的相关指导原则。
4.3.2.10稳定性试验数据应充分分析,并通过统计分析的方法确定NTIDs的有效期。应当将平均曲线的95%单侧或双侧置信限与认可标准的相交点所对应的时间点作为有效期。如果分析结果表明批次间的波动较小,可将数据合并进行整体分析评估。如果批次间的波动较大(P值≤0.25),则不能合并分析,有效期应当依据其中最短时间确定。
注:P值是用来判定假设检验结果的一个参数,P值≤025说明假设检验的两个批次间具有较大差异。P值的计算可参考统计分析方法的介绍。
4.4窄治疗指数药物质量标准的建立
4.4.1 NTIDs应建立科学合理的质量标准。
4.4.2根据NTIDs的特性,并结合体内外研究和临床使用监测数据,制定其质量标准。
4.4.3 NTIDs质量标准应当充分考虑活性成分的释放行为。
4.4.3.1应当关注溶出度或释放度等有可能与体内药物释放量相关的有效成分限度控制范围,特别是内控范围。
4.4.3.2按照《中华人民共和国药典》四部0931溶出度与释放度测定法要求,参考本文件的附录A,并基于药代动力学模型分析,充分考虑个体内血药浓度波动情况、药物稳定性情况和商业化生产控制需要,制定溶出度或释放度等合理限度指标。必要时应设置多个时间点或溶出(释放)度上限和下限来控制释药行为。
4.4.4 NTIDs质量标准应合理规定药物有效成分含量控制范围,特别是内控范围。
4.4.5按照《中华人民共和国药典》四部0941含量均匀度检查法和ICHQ6要求,见附录B,并基于药代动力学模型分析,充分考虑个体内血药浓度波动情况,药物稳定性情况和商业化生产控制要求,根据不同情况适度收紧NTIDs有效成分含量的控制范围。
4.4.6含量分析方法的准确度及精密度应当与限度要求相匹配。
4.4.7 NTIDs在制剂处方、工艺开发及相应的验证工作中应当重点评估含量均匀度。
4.4.7.1必要时建立内控标准或含量均匀度检查方法,并不得低于《中华人民共和国药典》四部0941含量均匀度检查法相应要求。
4.4.7.2产品质量回顾中应当重点关注批次间活性成分含量和/或含量均匀度、溶出(释放)度、稳定性等趋势分析,设定放行标准,及时发现异常趋势,开展调查和启动纠正预防措施,持续提升产品质量控制水平。
4.4.8应当对NTIDs开展持续性评价工作,跟踪分析疑似药物不良反应与药物质量的相关性,对药物质量定期进行回顾分析,持续更新和完善质量标准。
附录A
(规范性)
窄治疗指数药物溶出度与释放度试验方法的建立与验证
A.1概述
A.1.1本附录旨在为建立一个科学、耐用和具有适度区分力的溶出度或释放度试验方法提供技术指导,为溶出度或释放度分析方法验证提供通用性的规范。溶出度与释放度试验不仅是药物研发和质量控制的重要手段,而且在药物全生命周期质量管理中起到关键作用。
A.1.2溶出度或释放度试验分析方法验证应符合《中华人民共和国药典》四部9101分析方法验证指导原则的要求。溶出度或释放度的判定标准应符合《中华人民共和国药典》四部0931溶出度与释放度测定法。
A.1.3溶出度或释放度试验方法的建立应主要考虑药物本身的理化性质、溶出介质、溶出装置和分析步骤等方面的内容。
A.1.4良好的溶出度或释放度试验方法应具有区分处方、生产工艺和生物利用度等因素显着变化的能力。
A.1.5 NTIDs溶出度或释放度试验方法应当达到一定的精密度要求。
A.2适用范围
A.2.1本附录主要适用于口服固体制剂溶出度或释放度试验方法的建立和分析方法的验证。其他剂型可根据具体情况,选择合适的溶出装置和溶出介质,参考本附录建立溶出度或释放度试验方法。
A.2.2未列入《中华人民共和国药典》的其他溶出度或释放度方法,研究者可参考本附录和其他国家药典,建立与制剂相适应的溶出度或释放度试验方法。
A.3具体要求
A.3.1前期试验的准备
A.3.1.1前期试验准备应重点关注原料药溶解度和稳定性、溶出介质和体积以及溶出装置的选择。
A.3.1.2可根据原料药的溶解度等性质以及药物在体内的环境,选择多种溶出介质进行研究。应当关注缓冲液或酸的浓度对药物溶解度的影响。
A.3.1.3可使用介质模拟胃液和肠液的组成,用于模拟制剂在体内的溶出行为。
A.3.1.4应当确保药物在溶出介质中可以稳定存在。如活性成分的降解不可避免,且降解可形成稳定的降解物,应测定降解产物或测定降解产物与药物的总和。
A.3.1.5通常溶出介质体积应当保持在《中华人民共和国药典》各品种正文项下或相关标准规定的范围内,且应当满足漏槽条件。
A.3.1.6NTIDs的溶出度或释放度检查一般不采用表面活性剂。如确需使用,则应当首选十二烷基硫酸钠等常用表面活性剂。使用浓度应当选择达到漏槽条件所需的最低浓度,应关注不同来源的表面活性剂质量可能会有差异。
A.3.1.7应当根据药物处方设计和制剂的体外行为选择装置,首选《中华人民共和国药典》四部0931溶出度与释放度测定法规定的装置,见表A.1。
A.3.2试验方法的建立
A.3.2.1脱气
A.3.2.1.1常见的脱气方法有加热介质、过滤和短时间抽真空。
A.3.2.1.2含有表面活性剂的介质通常不需要脱气。
A.3.2.1.3通常采用减少溶出介质的表面张力降低溶解气体对溶出过程的影响。
A.3.2.2沉降篮
使用桨法测定时,沉降篮常用于调整漂浮剂型的浮力,保持供试品在溶出杯底部或避免供试品粘附溶出杯(如薄膜衣片等),沉降篮应当与剂型相适应。
A.3.2.3转速
A.3.2.3.1对于普通胶囊或片剂,篮法常用转速为50-100 r/min,桨法常用转速为50或75 r/min,小杯法常用转速为25~100 r/min。
A.3.2.3.2桨法和篮法通常不采用25-150 r/min以外的转速。
A.3.2.3.3流池法的标准流速为4、8和16 mL/min。选择其它流速应提供依据。
A.3.2.4取样时间点
A.3.2.4.1溶出取样时间点一般是基于对溶出曲线的数据分析来确定的。
A.3.2.4.2对于在is min内释放85%以上的药物,单点或崩解就可满足要求。
A.3.2.4.3对于在约30~45 min内才逐渐达到85%以上的药物,通常需要足够多的时间点来表征溶出行为。
A.3.2.4.4缓控释制剂通常至少选择三个时间点来确定体外释放曲线,并要求药物累积释放量不少于80%。
A.3.2.5观察
在方法建立过程中应重点关注以下现象:
a)颗粒在溶出杯内分布不均,如颗粒粘附在溶出杯壁上,在杯底有锥状堆积现象,颗粒漂浮于介质表面,薄膜衣片粘附在溶出杯上;
b)气泡在容器内、装置上或单片制剂上,仪器上的光泽也可能是空气的气泡,当评价介质需要脱气时,应记录观察的内容;
c)样品摇晃或者旋转,或溶出桨碰撞到样品;
d)试验结束,去掉搅拌设备,观察颗粒是否粘附于桨或篮内;
e)是否存在薄膜或类似的结构;
f)在溶出介质表面,是否存在大量的漂浮颗粒或块状物;
g)观察崩解的速度;
h)缓释包衣或肠溶包衣的复杂崩解;
I)样品是否位于溶出杯的中心还是偏离中心,如果偏离中心,是否发生了粘附;
j)胶囊壳完全溶出或片剂崩解所需的时间。
A.3.2.6取样
A.3.2.6.1取样的位置必须符合《中华人民共和国药典》四部0931溶出度与释放度测定法的规定。
A.3.2.6.2当转速较慢时,如50 r/nun的篮法,应当注意在容器中的同一位置取样。
A.3.2.6.3应当确定每一时间点是否进行补液,一般不推荐补液,但当不满足漏槽条件时,则必须进行补液。
A.3.2.6.4溶出介质抽出放回,在整个时间点用于计算的体积是一样的,但每一个样品取出的药物在计算时应当考虑进去。
A.3.2.7清洗
A.3.2.7.1应当重点评估每次试验之间的清洗是否充分。
A.3.2.7.2应当观察容器壁上是否有吸附的残留物。
A.3.2.8数据处理
A.3.2.8.1溶出结果通过累积溶出量和戚溶出速率表示。
A.3.2.8.2溶出度结果的计算见表A.2。
A.3.2.8.3溶出速率通常以指定试验时间的标示溶出百分数来表示。
A.3.2.9溶出度试验方法的评估
A.3.2.9.1溶出度试验方法由仪器、溶出介质和试验条件组成。
A.3.2.9.2溶出度试验方法应当灵敏度高、耐用性强、重复性好,可用于日常测定和方法转移。
A.3.2.9.3筛选可以显着提高方法区分力的溶出度试验条件,如浓度、转速和脱气等。具有良好区分力的溶出度方法,应当能够反映制剂的关键质量属性(如辅料比例、粒径大小、压力等)的变化。
A.4分析
分析包括样品处理和检测两个步骤
A.4.1样品处理
A.4.1.1取出样品后,需要进行过滤以除去未溶解的颗粒,必要时进一步处理样品,以符合检测要求。
A.4.1.2应当根据抽取溶出液的体积和需滤除微粒的量来选择滤膜尺寸,不推荐对样品进行离心。
A.4.1.3如需要稀释样品,一般使用溶出介质或流动相来稀释样品。
A.4.2检测
A.4.2.1溶出样品含量测定常用分光光度法或高效液相色谱法。
A.4.2.2分光光度计的比色皿光程一般为0.2-4cm。如果NTIDs的测定浓度较小时也可以使用光程较大的比色皿增加检测灵敏度,注意药物降解和辅料干扰。
A.4.2.3高效液相色谱法需考虑溶出介质对色谱峰的影响。如果目标峰响应值较低,溶剂的干扰可能会影响响应值的准确度和精确度。如果进样量较大则影响会更为明显。对照品溶液一般应用溶出介质稀释至方法线性范围内的浓度。在某些情况下,样品可以用流动相稀释以改善峰形。
A.4.2.4对照品和样品溶液应在线性浓度范围内并用相同波长测定。
A.5自动化
A.5.1试验的准备、取样、清洗建议采用自动化装置。
A.5.2介质一般通过稀释浓溶液来制备,应关注浓溶液的稳定性以及稀释液的均一性。
A.5.3取样使用取样针或者光纤取样针,需要进行充分的验证以保证取样针不对药物的溶出速率产生显着影响,取样方式通常包括程序取样和循环取样。
A.5.3.1程序取样体积应充分考虑管路、试管和其它设备的死体积。
A.5.3.2循环取样可以弃去也可保留取样点间的管路溶液,应当考虑死体积和剩余溶液的影响。多个取样时间点的取样消耗可用等体积的新鲜介质进行补液。
A.6验证
A.6.1溶出度方法的验证包括分析方法和溶出过程的验证。
A.6.2分析方法验证包括准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性,供试品溶液及对照品溶液的稳定性。
A.6.3溶出过程是指药物在溶出介质中的溶出和取样。溶出过程的验证主要是评估供试品溶液制备的精密度和耐用性。
A.6.4分析方法的验证和溶出过程的验证可以同时进行,验证研究应包含溶出曲线的时间点。
A.6.5根据研究阶段或预期数据的要求不同,验证参数和验证要求会有所不同。
A.7判定标准
A.7.1判定标准应当依据NTIDs特性,按照《中华人民共和国药典》四部0931溶出度与释放度测定法的要求制定。
A.7.2NTIDs溶出的试验结果应当与临床试验结果相关,溶出试验和判定标准应当能区分临床使用不可接受的批次。
A.7.3当制剂处方和工艺过程发生显着改变影响溶出度时,溶出度试验和判定标准也应当能区分这些改变。
A.7.4对于NTIDs,必要时应当设置多个时间点或溶出(释放)度上限来控制释药行为。
附录B
(规范性)
窄治疗指数药物含量及含量均匀度限度设定
B.1概述
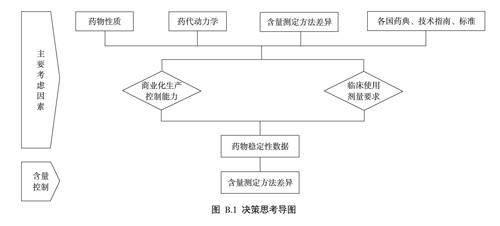
B.1.1药物的含量限度一般需要根据药物的性质、商业化生产控制能力、测定方法的准确度及临床使用剂量要求综合考虑,以制定一个合理可行、风险可控的质量控制范围。
B.1.2基于NTIDs在临床使用时,剂量或血药浓度有小的差异就可能导致严重治疗失败和/或药物不良反应的特点,因此应当严格控制限度范围。
B.1.2.1根据药代动力学模型分析,充分考虑个体内血药浓度变化情况、药物稳定性情况、产品批次间与批内的含量差异情况,参考各国药典相关品种标准及相应的技术指导原则,合理制定NTIDs的有效成分含量控制范围,详见图B.1。
B.1.2.2NTIDs仿制药含量限度一般应与参比制剂保持一致或收紧限度范围。
B.2窄治疗指数药物含量测定方法的选择和研究
B.2.1对于原料药和制剂含量测定,应选择准确度高、专属性强、能稳定反映产品质量的方法。
B.2.2含量测定和有关物质测定通常可以选用同一种方法(如高效液相色谱法)。
B.2.3如采用的方法是《中华人民共和国药典》收载方法,无需再验证但需要进行方法学确认。
B.2.4如采用的方法非《中华人民共和国药典》收载方法,则需要按照《中华人民共和国药典》和相关指导原则的技术要求进行方法学验证,并符合要求。
B.2.5应当关注NTIDs特殊剂型的含量控制标准,结合商业化生产能力来合理制定,确保含量可以持续符合标准要求。
B.2.6NTIDs注射剂需要过量灌装的增量应参考《中华人民共和国药典》四部0102注射剂的相关要求。
B.2.7NTIDs制剂处方和工艺开发及相应的验证工作中,建议评估含量均匀度标准制定的必要性,必要时建立相关检测方法。
8.2.8含量均匀度的限度,应当不低于《中华人民共和国药典》四部0941含量均匀度检查法的要求。



