禹雯怡 赵璐 孔宪琛 唐永平 万梅 邱建忠 袁荣涛
(1 青岛大学口腔医学院,山东 青岛 266003; 2 青岛大学附属青岛市市立医院口腔医学中心)
牙源性角化囊肿(OKC)是一种单囊或多囊的牙源性发育性囊肿[1],是临床上较常见的良性颌骨病损,约占牙源性囊肿的10%[2]。OKC可作为单独病变或痣样基底细胞癌综合征(NBCCS)的一部分出现,约5%的病例与NBCCS伴发[3]。OKC是颌骨最具争议性的病理实体之一,其特征为具有高生长潜力和复发倾向的局部侵袭性病变[4]。刮除术为目前临床上治疗OKC常用的方法,但术后复发率较高;开窗减压配合二期手术或颌骨节段性切除术可用于较大囊肿的治疗,但治疗周期较长,患者可能面临较大的手术创伤,并且术后并发症较多。揭示OKC发病机制是改进该病治疗方法的重要前提,然而其发病的起始因素及其分子机制目前仍不清楚。近年来研究表明,位于染色体9q22.3-q31上抑癌基因Patched(PTCH)突变及其引起的Sonic Hedgehog(Shh)信号通路的异常激活与OKC发病的关联程度最大[5]。OKC囊壁上皮组织中增殖细胞核抗原(PCNA)及Ki-67表达强度高于含牙囊肿和根端囊肿组织[6]。囊腔内正压力促进OKC上皮细胞炎性细胞因子白细胞介素-1α(IL-1α)的表达,可能参与了刺激破骨细胞的生成和激活病变周围骨吸收过程[7]。转录组测序技术(RNA-seq)就是采用高通量的测序技术进行测序分析,本研究对利用RNA-seq获得的数据进行分析,筛选与OKC发生、发展相关的差异表达基因(DEGs),并进行功能富集和调控网络分析,以期为OKC的靶向治疗寻找潜在作用靶点。
1 资料和方法
1.1 一般资料
2019年1月1日—12月31日在我院口腔颌面外科接受手术治疗,病理学证实为原发性非综合征型OKC患者3例,男2例,女1例;年龄38~67岁,平均54岁。所有患者在标本采集前均未接受任何其他治疗。本研究经我院医学伦理委员会批准通过(#2016-04-28-05),并征得患者书面知情同意。
1.2 样品收集和准备
收集3对囊壁和正常口腔黏膜组织(距离囊肿边缘≥2 cm)标本,离体后立即投入液氮中保存,用于转录组测序分析。
1.2.1RNA提取与检测 使用Trizol法提取组织RNA,Agilent 2100生物分析仪精确检测RNA完整性,Nanodrop超微量分光光度计检测RNA浓度和纯度。
1.2.2文库构建与质控 使用Illumina的NEBNext®UltraTM RNA Library Prep Kit生成测序文库。用附有Poly-T oligo(dT)的磁珠从总RNA中纯化mRNA,随后在高温下使用二价阳离子在NEBNext第一链合成反应缓冲液(5X)中将得到的mRNA进行片段化。然后使用随机六聚体引物和M-MuLV逆转录酶合成第一链cDNA,使用DNA聚合酶Ⅰ和RNase H合成第二链cDNA,随后使用AMPure XP beads进行cDNA纯化。纯化后的双链cDNA进行末端修复、加A尾并连接测序接头,采用AMPure XP系统对文库片段进行纯化,筛选长度为240 bp的cDNA片段。最后通过PCR富集方法得到cDNA文库。
在Agilent 2100系统上评估cDNA文库质量,insert size符合预期后采用实时荧光定量PCR(RT-qPCR)方法对文库有效浓度进行精准定量,其中文库的有效浓度应大于2 nmol/L。
1.2.3上机测序 文库质检完成后,根据目标下机数据量将不同的文库进行合并,随后采用Illumina Novaseq平台进行高通量测序,产生150 bp端读数。RNA-Seq相关性分析结果表明,样品间基因表达模式相似度质量评估合格,样本选择合理。
1.3 生物信息学分析
测序片段测得的图像数据经CASAVA碱基识别转化为序列数据reads,随后经过过滤得到的数据为Clean Data,并将此数据与指定的参考基因组进行序列比对。根据基因在不同样品或不同样品组中的表达量进行差异表达分析,以及DEGs功能注释、功能富集表达水平分析等,其中DEGs的筛选条件如下:P-value<0.01& |log2(FC)|>1,P-value为差异显着性P值,FC(Fold Change)为差异倍数。将3对OKC囊壁组织以及正常口腔黏膜组织的DEGs根据样本的FPKM值进行分层聚类分析,得到聚类热图。采用火山图表示基因整体分布情况,包括上调和下调2.0倍的DEGs及非DEGs。
2 结 果
2.1 基因转录本的差异分析
转录组测序结果显示,在3对OKC囊壁与正常口腔黏膜组织中总共检测到23 609个基因,其中DEGs 359个,上调者169个,下调者190个。
2.2 基因聚类分析
将3对样本的OKC囊壁和正常口腔黏膜组织的DEGs进行分层聚类分析,得到的聚类图中可见OKC和正常口腔黏膜组织能够完整分开(图1),表明组内样本具有较好的重复性,组间对应基因表达的差异性较大。图中横坐标为样品名称以及样品的聚类结果,纵坐标为DEGs及基因的聚类结果。颜色表示基因在样品中的表达量水平log10(gene+0.000 001),红色表示对应基因在对应组织中较高表达,绿色表示对应基因在对应组织中较低表达。
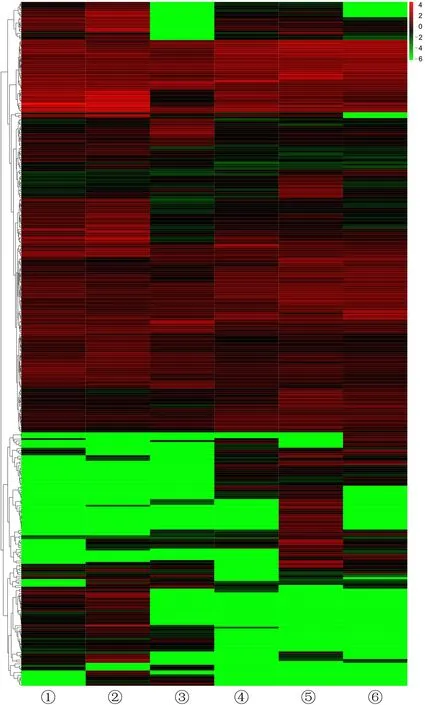
①~③:正常口腔黏膜组织,④~⑥:OKC囊壁组织图1 DEGs聚类热图
2.3 差异表达火山图分析
火山图展示了基因整体分布情况,OKC囊壁与正常口腔黏膜组织的测序结果包括上调和下调2.0倍的DEGs(P<0.01)以及非DEGs。横坐标绝对值越大,说明OKC囊壁与正常口腔黏膜组织间的表达量倍数差异越大;随着纵坐标值增大,差异表达显着性增加,筛选结果可信度也越高。见图2。
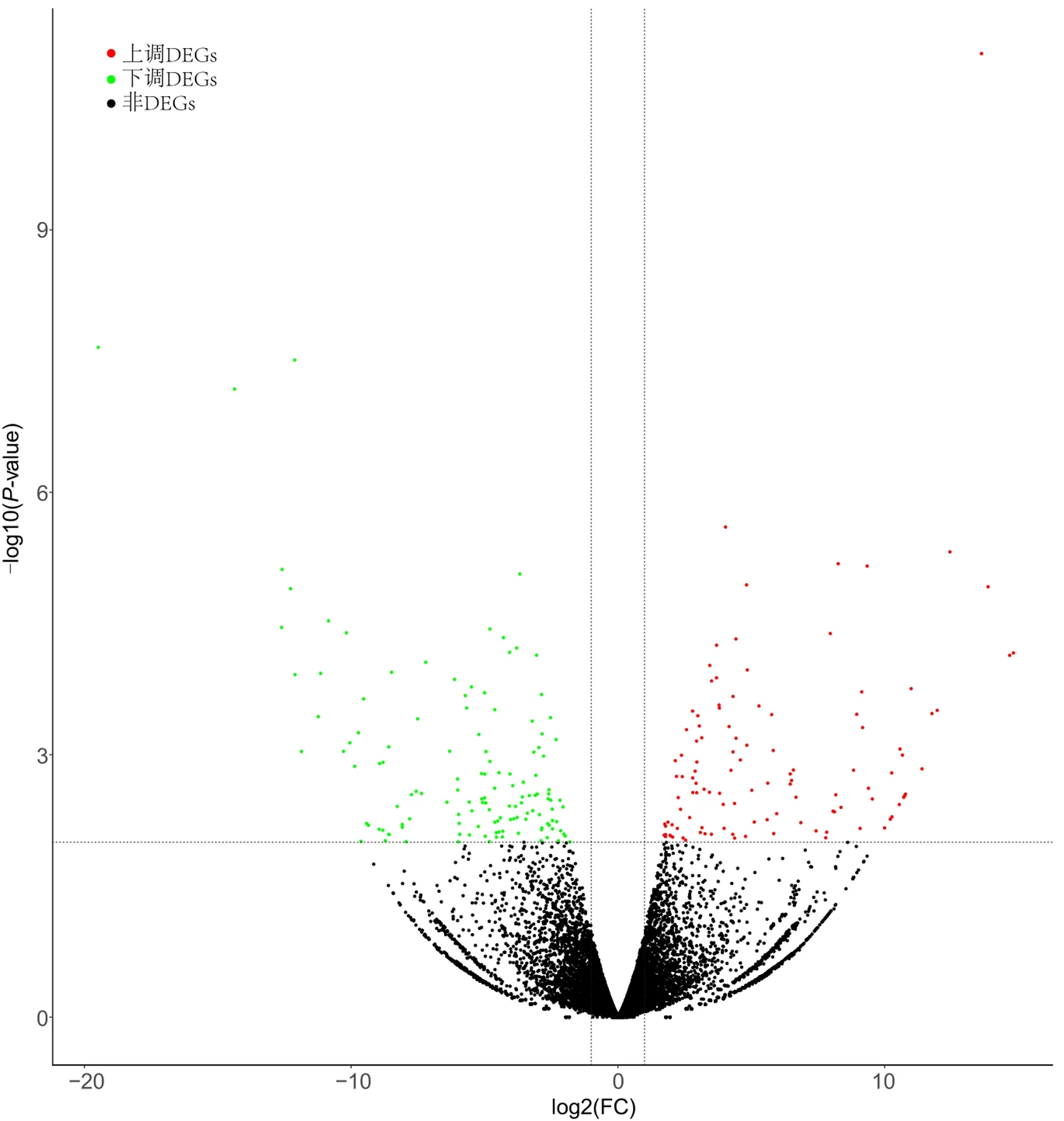
图2 DEGs火山图
2.4 DEGs功能富集分析
使用DAVID数据库对KEGG通路进行富集分析,结果显示OKC囊壁和正常口腔黏膜组织相比,注释在磷脂酰肌醇3-激酶/蛋白激酶B(PI3K-Akt)信号通路中的DEGs数目最多并且差异具有统计学意义(P<0.01)。见图3。图中展示了20个KEGG条目,横坐标表示DEGs中注释到该通路的基因比例与所有基因中注释到该通路的基因比例的比值。即富集因子越大,说明DEGs在该通路中的富集水平越显着。点的大小、形状、颜色分别代表注释在该通路中DEGs的数目、类型和q-value。
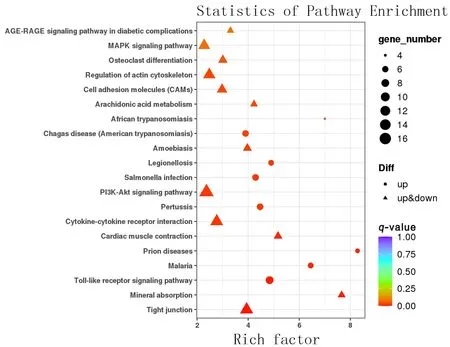
图3 DEGs的KEGG通路富集分析图
在DEGs的KEGG通路富集点图(图4)当中,GeneRatio表示注释在PI3K-Akt信号通路中的感兴趣基因占所有DEGs数的比例最大,在展示前10位中排位第1,15个DEGs。纵坐标为10个KEGG条目,横坐标表示注释在该条目中的感兴趣基因数与所有DEGs数的比值。点的大小表示DEGs数在该通路中的注释,点的颜色代表校正后的P值。

图4 DEGs的KEGG通路富集点图
3对样本测序结果富集到PI3K-Akt信号通路中的DEGs为15个,上调者8个,差异倍数由高至低分别为FGF5、IBSP、FGF9、SPP1、IL6、FN1、NR4A1、TNC。下调者7个,差异倍数由高至低分别为GYS2、PRLR、VEGFD、DDIT4L、FGFR3、LAMA3、YWHAB。
3 讨 论
基于深度测序的转录组分析提高了对DEGs、序列变异和新型融合转录本的检测,是继基因芯片技术后在基因检测技术方面的重大突破,因此在癌症研究中具有广阔的前景[8]。OKC的发生发展是一个复杂的生物学调控过程,可能存在多基因的突变及异常表达,本研究通过分析OKC囊壁与正常口腔黏膜组织间的基于RNA-seq获得的数据,以期筛选与OKC发生发展相关的关键基因及可能参与的信号通路。
OKC的发病机制尚未明确,一般认为OKC的组织来源为Serres上皮剩余。在牙齿发育完成后这些本应退化而残留于颌骨内的上皮剩余在不良刺激的诱导下可重获增殖活性,发生囊性化继而形成囊肿。还有学者认为其来源于口腔黏膜上皮的基底细胞增殖[7]。目前对OKC的研究包括遗传学因素、囊壁上皮细胞的增殖与凋亡、囊内液体正压力作用3个方面[9]。有研究证实PTCH1基因突变及其引起的Hh信号通路的异常激活可能是OKC发病的主要原因,高表达Shh、Smo以及Gli1的OKC可能与NBCCS相关[5]。经典Hh通路(PTCH1-SMO-GLI1)和非经典Hh通路(PTCH1细胞内环通过与CyclinD1结合调控细胞周期)均可能参与OKC病变发生过程[10]。OKC中存在Wnt信号通路相关基因的异常表达,Wnt5a、FZD3、MAPK10、PRKX、CAMK2A可能是通过调控细胞的增殖、分化,在OKC的发生、发展中起着重要作用[11]。表没食子儿茶素-3-没食子酸酯(EGCG)可能通过抑制WNT/JNK信号通路而抑制OKC角质形成细胞的增殖和诱导细胞凋亡[12]。最近一些关于牙源性肿瘤的研究表明,OPG/RANK/RANKL信号通路参与了OKC引起的骨吸收过程,开窗减压治疗可促进OKC附近骨的再生[13]。
本研究利用RNA-seq测序总共获得了359个DEGs,其中169个上调,190个下调,表明样本中基因组转录本具有显着的差异。KEGG通路富集分析表明,PI3K-Akt信号通路中的DEGs数目最多且差异具有统计学意义,检测到的15个DEGs可能为OKC潜在的生物标志物。PI3K-Akt信号转导通路通过影响下游多种效应分子的活化状态,参与肿瘤细胞的生存、增殖、周期进展、生长、迁移和血管生成等过程[14]。参与PI3K-Akt信号通路的调节因子主要为负调节因子,包括PTEN、SHIP2等。MiR-21通过PTEN/PI3K/Akt通路调控卵巢癌细胞的增殖和凋亡[15]。下调SHIP2基因,可通过PI3K-Akt通路增强喉鳞癌放射敏感性[16]。COX-2通过PI3K/Akt/NF-κB信号通路促进骨肉瘤MG-63细胞的上皮-间充质转化以及迁移过程[17]。富集至PI3K-Akt信号通路中的DEGs中变化倍数最显着的GYS2,其在肝细胞癌中的表达亦显着下调;敲低GYS2通过调节p53的表达,可促进细胞增殖和肿瘤生长,证明了GYS2通过与p53的负反馈回路抑制乙肝病毒相关性肝癌的生长[18]。在氧-葡萄糖剥夺/复氧模拟的脑缺血-再灌注损伤过程中,miR-145-5p可通过负调控FGF5促进神经细胞的凋亡和损伤[19]。
本研究中OKC囊壁组织中FGF9表达上调。研究显示,相比于癌旁组织,胃癌组织中FGF9基因表达上调,且FGF9通过激活ERK和Akt信号通路增强胃癌细胞抗凋亡能力[20]。新型长链非编码RNA-FAF通过PI3K/Akt信号通路上调FGF9基因表达,进而抑制缺血缺氧心肌细胞的凋亡[21]。沉默SPP1基因,可通过参与PI3K/Akt信号通路抑制舌癌的进展[22]。研究发现,IL-6的异常和过度分泌可能导致严重的炎症反应,PI3K/Akt通路的激活对IL-6反式信号介导的人血管内皮细胞促炎症反应至关重要[23]。天然二苯乙烯类化合物在体内具有抗炎作用,并且可能以PI3K/Akt依赖的方式下调IL-6的产生[24]。
Akt/mTOR通路在癌症中的作用众所周知,但很少有研究评估该信号通路在良性病变中的重要性。有研究发现Akt/mTOR通路在OKC组织中表达上调,该通路可能参与了病变的发生发展[25]。考虑到该信号通路在调控细胞增殖中的作用,可以解释为什么OKC表现出高水平的细胞增殖活性、侵袭性以及复发倾向。全基因组芯片分析OKC转录组特征,聚类分析结果提示有两个不同的分子亚型,Akt通路在一种亚型中被激活,MAP激酶通路在另一种亚型中被激活[26]。此外,PTCH1在两个亚型中表达均下调,表明其可能参与了OKC病变的进展。SHH/PTCH是一个已明确参与OKC发病机制的信号通路,在某些癌症中,SHH/PTCH可以通过Akt/mTOR通路促进细胞迁移、侵袭以及转移[27-28]。
总之,在本研究中筛选获得的DEGs,其中一些已经被认为是肿瘤的潜在生物标志物。研究OKC发病机制中所涉及的信号通路对于疾病的非手术治疗很重要。OKC中存在PI3K/Akt信号通路相关基因的差异表达,且注释在该信号通路中的感兴趣基因占所有DEGs数的比例最大,但目前的研究有一定的局限性,尚需要增加样本量及进一步的研究来阐明这些基因在OKC中的调控作用,并最终为OKC的靶向治疗寻找潜在作用靶点。



