乔虎军 王国祥 郝鑫
1 苏州大学体育学院(苏州 215021)
2 哈尔滨体育学院(哈尔滨 150008)
骨关节炎(osteoarthritis,OA)是以关节组织成份、结构和功能退行性改变为特征的慢性关节疾病,主要涉及关节软骨损伤并累及软骨下骨和周围结构(例如软骨下骨病变、骨赘和滑膜炎症等)。OA的发病部位主要包括膝关节、髋关节、手指关节、颈部、肩关节、肘关节、脊椎关节以及容易被忽视的踝关节和足等,通常伴有疼痛、肿胀和关节僵硬等。根据统计,全球已经有超过3.6亿人口患有OA[1],并呈现出一定的性别和年龄特点。美国的一项数据显示OA发生的概率和年龄趋势成正相关,并且提示性别可能成为一个危险因素,超过45岁的女性有着较高的膝骨关节炎(knee osteoarthritis,KOA)发病率[2]。超过60岁以后,受OA困扰的女性更是将近男性的两倍。作为发达国家主要的致残疾病之一,OA患者中有80%存在运动局限性,25%患有残疾[3]。
导致OA的因素是多方面的,例如遗传或基因突变等先天因素,体重增加引起的机械负荷过大、疾病引起的继发性炎症反应、过量体育运动或机械撞击引起的创伤、年老发生的软骨退行性改变等后天因素[4,5]。虽然这些因素在OA发生和发展过程中的影响机制已经开始被探索和认识,但仍缺乏有效的干预措施来阻止或减缓OA的发展进程。研究表明,转化生长因子β(transforming growth factor-β,TGF-β)/Smad信号通路参与早期软骨形成,调节软骨细胞内稳态,影响软骨细胞表型特征。TGF-β/Smad信号通路与OA的病理变化有着密切的联系。充分理解TGF-β/Smad信号通路的分子机制,有助于探索治疗OA的潜在靶点。因此,本文根据国内外相关研究对TGF-β/Smad信号通路与软骨细胞以及OA的关系进行综述。
1 TGF-β/Smad信号通路
1.1 TGF-β 及其受体
TGF-β属于TGF-β超家族成员,参与细胞增殖、识别、分化和凋亡等过程,并在哺乳动物早期发育和调节免疫应答过程中起到一定的调控作用。TGF-β在人类中有三种结构相似的亚型(TGF-β1、TGF-β2和TGF-β 3),它们拥有84%~92%的高度同源性,并被不同的基因所编码。首先,TGF-β基因编码形成前体分子(perpro-TGF-β),其主要由隐性相关肽(LAP)、成熟的TGF-β蛋白和一个信号肽等部分构成。信号肽在内质网中被切除后形成无活性的pro-TGF-β。随后通过弗林蛋白酶在反式高尔基体中进一步加工,最终成熟的TGF-β从LAP中分离出来。但这两种多肽仍以非共价键的形式形成复合体(small latency complex,SLC),抑制成熟TGF-β的活性。然后,TGF-β可以通过蛋白酶水解(纤溶酶,MMP-2,MMP-9)、活性氧化物质(ROS)刺激、血小板反应蛋白-1(TSP-1)与LAP相互作用、酸性环境刺激或者与整合素的相互作用等方式而激活[6]。
TGF-β超家族有三种受体,分别为Ⅰ型(ALK1-7)、Ⅱ型(TβRⅡ,ActRⅡ,ActRⅡb,BMPRⅡ和MISRⅡ)和Ⅲ型共同受体(CD109、内皮因子、β-蛋白聚糖)。其中Ⅰ型受体和Ⅱ型受体都是单通道跨膜受体,是含有特殊丝氨酸/苏氨酸激酶结构域特征的糖蛋白。信号传导过程中,Ⅰ型受体不能直接和配体分子结合,但可以在配体分子存在下和Ⅱ型受体形成亲和力比较高的受体复合物。Ⅱ型受体自身具有多个磷酸位点,具有持续的激酶活性,因而可以在两种受体结合后激活Ⅰ型受体,进而启动下游通路。Ⅲ型共同受体中的β-蛋白聚糖是一种没有蛋白激酶活性的蛋白多糖,可以增加配体和另外两种受体的亲和力。此外,作为TGF-β超家族的共同受体,β-蛋白聚糖参与TGF-β超家族配体的运输,是信号输出的决定因素[7]。
1.2 Smad
Smad蛋白处于TGF-β/Smad信号通路的核心位置,在信号传导以及调控下游目标基因转录过程中扮演重要角色。已知的哺乳动物Smad蛋白家族包括Smad1、Smad2、Smad3、Smad4、Smad5、Smad6、Smad7和Smad8(有报道称为Smad9)。根据其特有的构造和功能,将Smad蛋白分为R-Smad,Co-Smad和Ⅰ-Smad。这些蛋白在TGF-β超家族发起的Smad信号通路中发挥不同的功能。R-Smad包括Smad1、Smad2、Smad3、Smad5和Smad8,统称为受体调节Smad蛋白,可以和具有激酶活性的Ⅰ型受体相互作用而激活。Smad4被称为公用型Co-Smad蛋白,可以为所有的R-Smad提供辅助,其主要功能就是协助信号分子进入细胞核。Ⅰ-Smad是一类对TGF-β超家族信号传导起到抑制作用的Smad蛋白,主要包括Smad6和Smad7。其可以干扰R-Smad同受体或Smad4的结合,进而对TGF-β超家族Smad依赖性信号途径发挥调节或抑制作用[8]。
1.3 TGF-β /Smad信号通路的分子机制
如图1,TGF-β/Smad信号通路主要是TGF-β信号分子,细胞膜表面Ⅰ型和Ⅱ型丝氨酸/苏氨酸跨膜受体以及细胞内效应物质Smad通过特异性膜结合的方式传导。研究表明,Ⅰ型受体ALK决定何种R-Smad被磷酸化,是信号传递的重要环节。该特异性主要由Ⅰ型受体β-折叠结构中β4和β5之间的环和R-Smad蛋白中L3环之间的相容性决定。典型的TGF-β/Smad信号路径为TGF-β/ALK5/Smad2,3,然而越来越多的研究发现TGF-β还可以通过ALK1/Smad1,5,8途径传导信号。两条信号通路分别在软骨细胞终末分化过程中发挥相反作用[9]。研究证实,TGF-β/Smad2,3信号传导有利于维持软骨细胞内稳态,而TGF-β/Smad1,5,8信号通路则会引起软骨细胞肥大[5,10]。
如图1所示,活化后的TGF-β先后和两种受体结合,并在细胞膜表面形成异源四聚体受体复合物。然后,具有持续性激酶活性的TβRII在受体复合物中磷酸化ALK1或ALK5,使ALK1或ALK5活性得到激活。受体复合物在细胞膜表面通过clathrin蛋白介导的内吞作用而进入细胞质[11]。进入细胞后,活化的Ⅰ型受体(ALK5或ALK1)可以和Smad2,3或Smad1,5,8短时间结合,并使其磷酸化。接下来,Smad2,3或Smad1,5,8从受体复合物中分离并和Co-Smad相互作用,形成具有功能的三聚体并进入细胞核内[12]。然后,三聚体复合物和带有转录因子的目标DNA相互作用并招募不同的复合物,进而激活不同的基因组[13]。
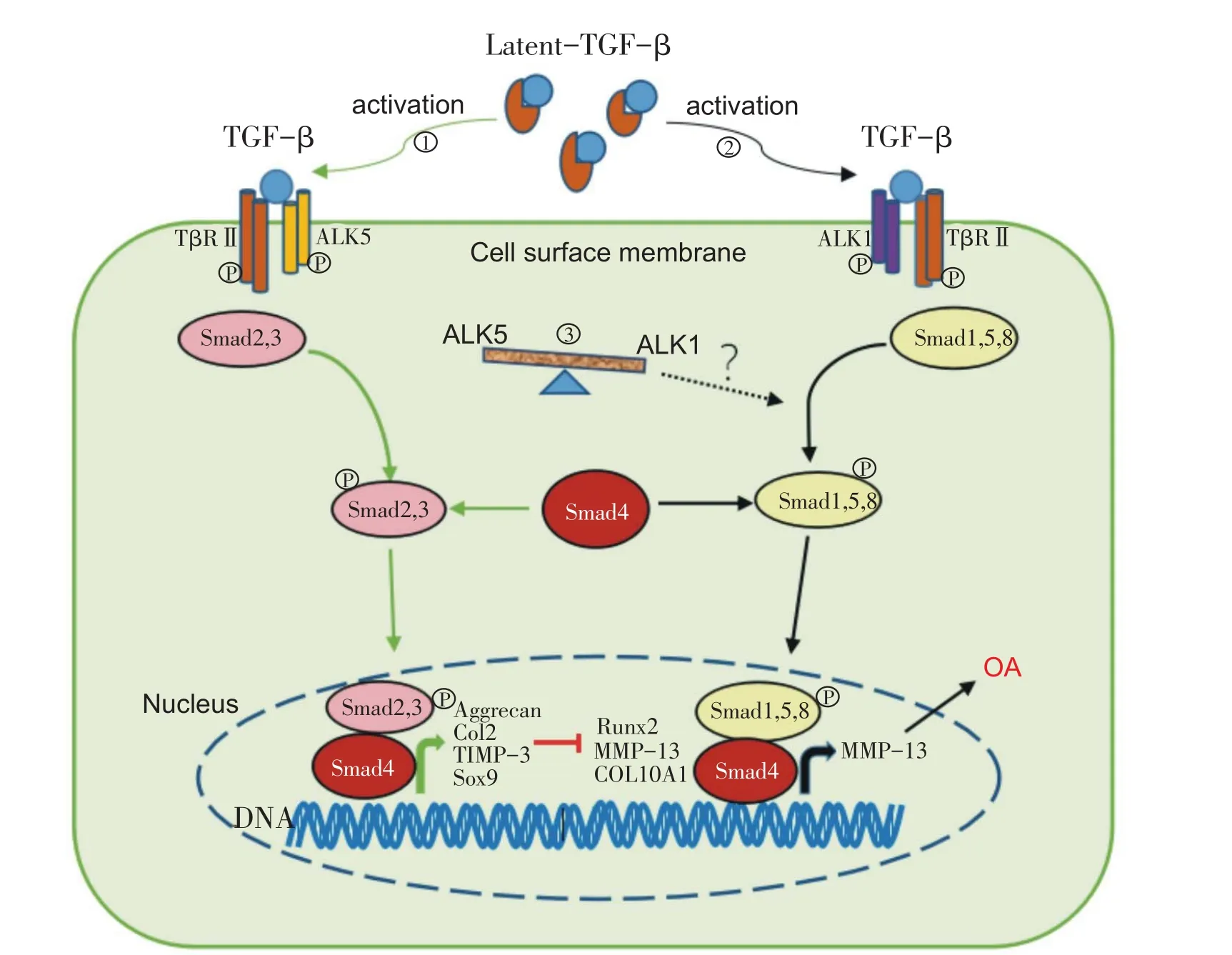
图1 TGF-β介导的Smad信号路径
2 TGF-β/Smad信号通路与软骨细胞
2.1 软骨细胞形成
软骨细胞可以分为早期软骨细胞形成阶段和软骨细胞肥大阶段:早期以转录因子Sox9上调,软骨细胞外基质Ⅱ型胶原蛋白和聚集蛋白多糖沉积为特征;肥大阶段则以软骨细胞体积增大,软骨细胞外基质重塑、终末分化标志物成骨转录因子Runx2、肌细胞促进因子MEF2C、MMP-13、X型胶原蛋白、碱性磷酸酶(ALP)以及印度刺猬蛋白(Ihh)等增加为特征[14-16]。
软骨细胞形成于胚胎发育期的间充质细胞,同时伴有环磷酸腺苷(cAMP)、TGF-β、纤连蛋白、神经细胞粘附分子(N-CAM)、N-钙粘蛋白等物质增加[14]。间充质细胞先后分化为骨祖细胞和成软骨细胞(此时胞外基质和纤维不断形成),最后形成两种主要的软骨细胞,即关节软骨细胞和生长板软骨细胞。早期软骨细胞形成阶段,关节软骨细胞和生长板软骨细胞拥有共同的细胞表型特征。然而,两种细胞什么时候开始形成特有的表型特征并不十分清楚。它们最显着的区别在于,关节软骨细胞不会像生长板软骨细胞一样进入肥大前状态。即关节软骨细胞终末分化被抑制形成永久性透明软骨。然而,骨关节炎发展期间,关节软骨中的软骨细胞表型发生变化,形成类似于仅在终末分化期的软骨细胞表型。
TGF-β/Smad2,3信号通路可以促进软骨细胞Ⅱ型胶原蛋白和聚集蛋白多糖的形成[17,18]。事实证明此信号通路在软骨细胞扩增以及形成软骨样组织过程中发挥重要作用。其潜在机制是TGF-β/Smad2,3信号通路上调转录因子Sox9,增加软骨细胞外基质分子。在骨髓间充质干细胞形成软骨的研究过程中发现,Smad3和Smad4比Smad2发挥更为重要的作用[19]。
Sox9是早期软骨细胞、生长板软骨细胞和关节软骨细胞形成过程中的重要转录因子,在软骨细胞分化的不同阶段发挥不同的作用[20]。大量研究表明,Sox9可以促进软骨特异性基质分子(Ⅱ型胶原蛋白和聚集蛋白多糖)的形成。染色体免疫共沉淀测序分析发现,在早期软骨细胞形成过程中(肥大阶段前),Sox9的目标基因几乎包括了所有的软骨细胞外基质分子以及众多软骨特异性调节因子[21,22]。事实证明,Sox9和Sox5/Sox6相互协作,通过刺激超级增强子促进软骨细胞外基质沉积,进而驱动软骨形成[21,23]。研究发现TGF-β1可以促进Sox9mRNA和Sox9蛋白表达[24,25],而且可以分别通过两条独立的信号通路p38和Smad2,3调节Sox9的磷酸化和稳定[26]。组蛋白去甲基化酶(KDM4B)减少会降低Sox9启动子和Smad3的结合,推测组蛋白去甲基化酶(KDM4B)是Smad依赖性激活Sox9不可缺少的一部分[27]。
2.2 软骨细胞内稳态
软骨细胞形成后,TGF-β/Smad2,3信号通路有助于维持软骨细胞内稳态,抑制软骨细胞肥大标志物的产生。软骨细胞终末分化阶段受到转录因子Runx2的严格调控。Sox9可以通过与Runx2直接作用或通过介导NKx3.2/BaPx1抑制Runx2。此外,Sox9通过与GLI2/3结合,影响X型胶原蛋白基因上游的增强子,抑制X胶原蛋白的产生[28]。通过重组腺相关病毒感染,实现骨髓间充质干细胞TGF-β和Sox9共表达发现,Ⅱ型胶原蛋白和聚集蛋白多糖显着增加,并伴有明显的细胞扩增、DNA合成以及软骨细胞形成活动,同时肥大样标志物Ⅰ型和X型胶原蛋白降低[29]。
体外培养的原代牛软骨细胞,经过BMP9处理后引起Smad1,5磷酸化且伴有下游基因bAlpl表达,并在1周内形成肥大的软骨细胞。此现象在少量TGF-β1存在下得到抑制,表明TGF-β1在维持软骨细胞表型过程中具有重要作用[30]。通过腺病毒转染兔关节软骨细胞的方式实现TGF-β1过表达,聚集蛋白多糖和Ⅱ型胶原蛋白增加,Ⅰ型胶原蛋白减少,软骨细胞形态也从长梭形恢复成圆形或椭圆型[31]。此外,TGF-β1过表达显着增加Sox9 mRNA,并且抑制肥大标记物X型胶原基因COL10A1和矿化标记物MMP-13的产生。
基质金属蛋白酶抑制因子-3(tissue inhibitor of metalloproteinase-3,TIMP-3)对软骨起到一定的保护作用,可以抑制MMP-1、MMP-2、MMP-3、MMP-9及MMP-13等基质金属蛋白酶对软骨基质的降解。TGF-β/Smad2,3信号通路可以刺激软骨细胞TIMP-3的表达[32]。这一结论进一步被证实,同时强调细胞外调节蛋白 激 酶1(extracellular signal-regulated kinase 1,ERK1)在TGF-β诱导TIMP-3表达过程中发挥重要的调节作用[33]。
ALK1和ALK 5的比值升高会造成OA样软骨细胞表型。推测TGF-β/Smad信号通路由Smad2,3过渡到以Smad1,5,8传导为主,进而形成一种类似于发生在骨关节炎以及老化软骨细胞中的终末分化软骨细胞表型。有关研究表明,ALK5的减少会导致软骨细胞内稳态受到破坏从而引起软骨细胞终末分化[15]。OA和老龄模型鼠膝关节软骨中ALK1/ALK 5比值均升高,鼠软骨细胞中增加ALK1或抑制ALK5均引起MMP-13表达增加,造成OA样软骨细胞表型[34]。通过诱导小鼠软骨ALK5特异性缺失,导致其产生OA样软骨细胞表型,并伴有软骨退化、滑膜增生和骨赘产生等现象。同时,软骨细胞中合成代谢和分解代谢相关因子表达失衡,软骨细胞过度凋亡[35]。此外,敲除小鼠ALK5基因,PRG4mRNA和蛋白水平均降低。
人软骨细胞中,ALK5可以促进TGF-β1刺激下PAI-1、纤连蛋白和Ⅱ型胶原蛋白等物质的产生,ALK1发挥相反作用[36]。同时发现,TGF-β1可引起Smad1,5磷酸化,条件是需要ALK1和ALK5共同参与。ALK5可能在Smad1,5磷酸化过程中扮演更为重要的角色。通过LDN-193189抑制ALK1,2,3并没有影响TGF-β1诱导的Smad2和Smad1,5磷酸化或下游靶基因表达。相反,通过SB-505124抑制ALK4,5,7则阻碍了TGF-β 1刺激下Smad2和Smad1,5的磷酸化以及下游的转录活动[37]。该研究表明,ALK4,5,7在Smad1,5和Smad2,3磷酸化过程中都发挥重要作用。结果同时表明,Smad1,5的磷酸化可能并不需要ALK1,2,3的参与,这和以往的研究结论ALK1,2,3,6可以磷酸化Smad1,5,8相矛盾,确切结论有待更多的研究来验证。
综上所述,TGF-β/Smad2,3信号通路可以增加软骨细胞Sox9、TIMP-3、Ⅱ型胶原蛋白和聚集蛋白多糖等物质,并对Runx2、X型胶原蛋白和MMP-13等物质起到一定抑制作用。当ALK1/ALK 5比值升高,TGF-β/Smad1,5,8信号通路发挥主导作用,MMP-13表达增加,PAI-1、纤连蛋白和Ⅱ型胶原蛋白等物质降低,导致OA样软骨细胞表型。ALK 5可能在TGF-β发起的Smad2,3和Smad1,5,8两条信号通路中都发挥作用,其具体机制仍不清楚。
3 TGF-β/Smad信号通路与OA
3.1 基因突变与OA
随着全基因组关联分析的快速发展,TGF-β/Smad信号通路中相关分子的遗传突变与OA之间的联系已有报道。有研究发现,TGF-β1 rs1982073C>T变异可能会增加骨折、骨质疏松和OA的易感性[38]。此外,有报道称TGF-β1突变可能会引起恩格尔曼综合症[39,40]。恩格尔曼综合症是一种以渐进性皮质增厚和长骨硬化为特征的进行性骨干发育不良疾病。骨硬化被认为与OA的发展有关。
相比TGF-β,Smad3基因突变与OA的联系得到了更多报道。Smad3由位于15号染色体长臂2区1带至2带区间的Smad3基因控制,包括9个外显子和8个内含子。通过对527名欧洲患者分析发现,Smad3基因内含子区域的单核苷酸多态性与膝关节骨关节炎和髋关节骨关节炎发生率有关[41]。国内的一项研究也发现,Smad3基因遗传突变可能诱发膝关节骨关节炎和手部骨关节炎[42]。Smad3基因rs12901499位点,GA、GG基因表型以及变异G和膝关节骨关节炎的发生率升高有显着相关性[43]。类似的研究结果在髋关节骨关节炎患者中得到证实[44]。此外,通过限制性片段长度多态性聚合酶链反应(PCR-RFLP)技术分析112名膝关节骨关节炎患者和120名健康对照者基因型发现,OA组Smad3rs12102171 TT型和rs2289263 GG型要明显高于健康对照组[45]。
动脉瘤-骨关节炎综合征(AOS)被描述为一种由Smad3基因突变引起的常染色体显性遗传病,其表现为动脉瘤、动脉分层和动脉迂曲,并有早发性骨关节炎,轻度颅面、骨骼和皮肤异常等特征[46]。通过分析8个AOS家庭(8种Smad3基因突变)共计45名患者的临床表现发现,在接受放射学检测的26位AOS患者中有25位被确认为OA[47]。到目前为止,已经在Smad3基因中发现了29种不同的外显子突变[48]。突变位点遍布整个基因,尤其在外显子6区域存在较高的突变率。大多数突变发生在Smad3基因MH2结构域中,该变化将阻碍Smad3和Smad4的寡聚化以及Smad依赖性转录激活。然而,也有部分研究发现Smad3基因突变携带者(AOS患者)没有出现OA症状[49,50]。由于突变是在心脏疾病背景下研究的,未发现OA症状有可能是源于不完整的诊断方法。以上发现表明,TGF-β/Smad信号通路中相关分子的遗传变异与OA的发展密切相关。
3.2 TGF-β /Smad信号通路与OA软骨损伤治疗研究
TGF-β/Smad信号通路受到干扰可能诱发软骨损伤。软骨细胞中TGF-β1受到microRNA-483-5p抑制后,聚集蛋白多糖和Ⅱ型胶原蛋白mRNA含量都降低,MMP-13和Runx2mRNA水平均升高[51]。Ⅱ型TGF-β受体对维持正常软骨细胞表型不可缺少[52],通过诱导Ⅱ型受体基因缺失导致关节软骨中Runx2,Mmp13和Adamts5表达上调以及基因缺失小鼠渐进的OA发育[53]。特异性敲除小鼠Ⅰ型受体ALK5得到类似的结果[38]:软骨分解代谢因子过度生成,合成代谢因子表达降低,发生OA样软骨损伤。同样,Smad3基因敲除(Smad3-/-)小鼠也逐渐发展为终末期OA表型[54]。以上研究结果提示如何恢复TGF-β/Smad信号通路平衡可能成为减缓OA软骨损伤的关键。TGF-β/Smad信号分子作为OA软骨损伤治疗的重要研究内容,研究重点主要集中在TGF-β。收集OA晚期患者受损半月板,对提取出的细胞进行TGF-β3刺激,结果显示Runx2显着下降,Smad2,3、P-Smad2、Sox9明显增加[55]。
通过诱导TGF-β表达促进OA软骨细胞的合成代谢,增加Ⅱ型胶原蛋白和蛋白多糖的产生并抑制X型胶原蛋白形成[56]。同样,诱导TGF-β表达促进体外培养的OA软骨重塑,增加TIMPs表达,抑制OA软骨中X胶原蛋白、MMP-13、甲状旁腺激素相关肽(PTHrP)和β-连环蛋白(β-catenin)等物质的形成。此外,ALK1和ALK5的表达均增加且保持良好的ALK1/ALK5平衡。
采用软骨细胞介导的TGF-β1基因疗法(TG-C)对动物(兔、羊)OA模型进行干预治疗,特定剂量下Ⅱ型胶原蛋白增加并发现再生软骨[57]。TG-C初期临床实验发现,注射软骨细胞12个月后,一位患者可见新生软骨组织,12位患者在关节炎指数、关节僵硬评分、疼痛感和关节活动方面均有不同程度改善[58]。
TGF-β1可以刺激软骨细胞浅区蛋白(SZP)的表达[59,60]。SZP又称为润华素或蛋白多糖4(PRG4),这些蛋白由表面区软骨细胞和滑膜分泌,起到润滑关节的作用,所以SZP在软骨表面的累积对关节稳态十分重要。免疫印记法和酶联免疫吸附实验证实,浅表层软骨细胞表面的粘多糖影响TGF-β1对SZP积累的刺激[61]。抑制Smad3磷酸化可以抑制TGF-β1诱导的SZP剪接作用以及SZP蛋白的表达,说明SZP可变剪接以及蛋白表达受到TGF-β/Smad2,3信号通路调控[59]。
3.3 增强TGF-β /Smad信号传导可能加重OA
鉴于TGF-β/Smad信号通路在维持正常软骨细胞功能过程中发挥的重要作用,给予外源性TGF-β治疗OA软骨损伤的报道并不少见。然而,研究者发现TGF-β表达过高也可能会产生一定的负面作用。增强TGF-β/Smad信号传导可能诱发与OA有关的软骨损伤、软骨下骨病变和滑膜纤维化等病理变化。有学者发现OA关节软骨中TGF-β1/Smad2,3信号增强,通过敲除TβRⅡ抑制该信号通路则会使软骨损伤减轻[62,63]。此外,通过抑制不同OA动物模型中TGF-β1活性可以减轻OA病理特征并减缓关节软骨的退化[64]。另外一项研究也发现,TGF-β1过多表达会削弱TGF-β 1/Smad信号通路对OA的缓解作用,抑制TGF-β1活性则减轻软骨损伤[65]。此外,通过研究自发性OA模型发现软骨退行性病变可能与TGF-β/Smad信号通路改变有关[66]。结果显示随着年龄的增长,关节软骨pS-mad2,3水平下降,而pSmad1,5,8水平上升。衰老机制可能调控TGF-β/Smad信号通路,使其从Smad2,3偏向Smad1,5,8通路[67],提示过度的TGF-β活动可能加速软骨的退化。
随着近年来对OA病理机制的不断研究,学者们开始关注软骨下骨和OA的联系,并将软骨和软骨下骨视为一个整体的功能单位。传统观点认为紧邻软骨下骨板的钙化层是两者不能通过的屏障,事实证明软骨和软骨下骨间存在相互串扰,软骨下骨板上的孔隙可能成为串扰的路径[68]。尤其是在发生OA后,软骨下骨板上的孔隙增加[69,70],这种串扰可能会更加强烈。推测OA发生后,滑液中的代谢因子异常,并通过软骨下骨板的孔隙相互渗透,造成软骨和软骨下骨间的相互作用[68]。关节中异常的TGF-β表达可能同时影响关节软骨和软骨下骨,研究指出活化的TGF-β可以促进间充质干细胞向骨重塑位点转移,耦合骨吸收和骨形成[71]。OA发生过程中软骨下骨异常重塑可能与此有关。研究认为TGF-β1会产生类骨样小岛和异常骨组织并改变软骨下骨微结构[64]。一项关于胶原诱导性关节炎的研究证实,软骨下骨病理变化与异常的TGF-β1活性有关,而且在软骨下骨中发现较高的pSmad2,3水平[72]。推测TGF-β/Smad信号通路可能在OA软骨下骨病变中发挥一定作用。
持续的TGF-β信号活动可能导致多种组织的纤维化,例如肝脏、肺部、心脏、肾脏、骨和皮肤等[71]。同样,TGF-β过度表达可能引起滑膜纤维化[73]。赖氨酸羟化酶2b(LH2)与骨关节炎纤维化成正相关,研究发现TGF-β可以通过ALK5/Smad2,3信号传导诱导PLOD2/LH2产生。Blaney等向小鼠关节注射介导TGF-β的腺病毒载体2周后,滑膜宽度是对照组的2.5倍,Ⅰ型骨胶原的表达量是对照组的2.45倍,这些指标说明TGF-β导致滑膜纤维化[74]。同样是通过腺病毒载体实现关节TGF-β1过表达,Watson等发现裸鼠膝关节出现了严重的纤维化[75]。提示TGF-β1可能是关节纤维化的诱导因子。
此外,有研究提出,软骨细胞中TGF-β通过ALK5/Smad2,3依赖性信号传导方式刺激神经营养因子(NGF)表达。OA疼痛和NGF成正相关,这可能揭示了OA中潜在的非炎性疼痛的来源[76]。
3.4 TGF-β /Smad信号通路可作为治疗OA的潜在靶标
综上所述,增强的TGF-β/Smad信号传导可能促进OA的发展,如何抑制其信号传导可能成为治疗OA的重要内容。为此,众多研究者进行了不断的探索。Xue等通过慢病毒载体介导shRNA敲除Smad4,从而抑制TGF-β/Smad信号传导[77]。体内和体外研究结果显示,敲除Smad4可以抑制波形蛋白、α-SMA、collagenⅠ、collagen III、Lama1和Timp1等纤维化标志物的产生。Zhen GH等通过多种手段干扰TGF-β/Smad信号传导,不同程度地减轻了ACLT模型鼠的OA症状[64]。软骨下骨局部进行TGF-β1中和抗体处理后,减轻了异常软骨下骨形成和关节软骨退化;关节内注射TβRI抑制剂后,稳定软骨下骨结构,减轻软骨退变;诱导性敲除TβRII后,软骨蛋白多糖丢失减少,软骨钙化减轻,MMP-13和collagen X得到抑制。Chen等证实通过特异性基因敲除TβRII或使用TβRII抑制剂均可以减轻OA(DMM模型)引起的软骨退变[62]。Fang等发现成年小鼠退行性踝软骨中TGF-β1和pSmad2,3水平升高,TβRII基因敲除可以减轻踝软骨退变[63]。此外,Blaney Davidson等[74]通过向小鼠膝关节注射IL-1的方式诱导软骨损伤,并注射表达TGF-β和Smad7的腺病毒载体进行干预。结果显示,Smad7可以减轻TGF-β引起的滑膜纤维化,同时不影响TGF-β对软骨的修复。
综上,TGF-β/Smad信号通路和OA的具体关系仍然没有定论。全基因组分析证明TGF-β和Smad3基因突变可能诱发与OA相关的疾病。干扰TGF-β/Smad信号传导诱发OA的研究不在少数。然而,随着研究的深入,部分学者得出了相矛盾的结论。例如,TGF-β/Smad信号通路可能引起软骨损伤、软骨下骨病变以及滑膜纤维化等OA症状。而且,研究者尝试多种手段以阻断TGF-β/Smad信号传导的方式治疗OA,并得到了较理想的治疗效果。因此,TGF-β/Smad信号通路和OA的具体关系仍需要更多的实证研究来确定。
4 小结
目前研究表明,TGF-β/Smad信号通路参与早期软骨细胞形成,并在维持软骨细胞内稳态过程中发挥重要作用。TGF-β信号分子I型受体ALK1和ALK5的比值升高会影响软骨细胞内稳态,甚至造成OA样软骨细胞表型。TGF-β/Smad信号通路异常可能诱发OA样软骨损伤,加剧OA症状。然而,也有证据表明过强的TGF-β/Smad信号传导可能诱发软骨损伤、软骨下骨病变和滑膜纤维化等疾病。笔者推测TGF-β/Smad信号通路具有一定的时间和空间特征(不同组织),在不同时间和空间产生不同的生物学效果。因此,充分理解OA的发病机制,掌握OA后关节内不同组织中TGF-β/Smad信号通路相关分子的变化规律并恢复其上下游分子的平衡可能为治疗OA提供一定思路。随着全基因组关联分析的发展,可筛选与早发性OA有关的遗传变异(例如Smad3),为早期发现易感患者,理解发病机制,预防和治疗OA提供依据。TGF-β/Smad信号通路和OA的关系仍没有十分明确,需要更多的研究来阐明。



