赵阳飞,于洋欢,王金明,张建海,孙子龙,牛瑞燕,王俊东
(山西农业大学动物医学学院,太谷 030801)
由于地理因素和环境因素的影响,氟广泛存在于人畜生活的水和食物中。长期过量的氟暴露能够导致人畜氟中毒。畜禽氟中毒导致繁殖率、生产性能、使用价值降低,使其成为威胁我国畜牧业发展的潜在因素[1-2]。肝作为机体重要的解毒和代谢器官,前期研究氟能够导致肝结构和功能的损伤[3]。肝损伤能够导致机体物质代谢紊乱和有害物质的蓄积,进而影响畜禽的生产性能和动物性食品安全。因此,深入探究氟致肝损伤的致病机理寻找可能的治疗靶点对于维持畜牧业的可持续发展和动物性食品安全具有重要意义。
IL-17作为Th17细胞分泌的促炎因子,其家族包含IL17A~F。IL-17家族中IL-17A含量最多,且生物活性最强,通常所说的IL-17即IL-17A[4]。前期研究发现氟暴露后肝中IL-17A显着升高。IL-17A的高表达将导致中性粒细胞的募集和免疫细胞的慢性浸润[5]。同时,IL-17A可通过IL-17A受体通路促进细胞因子的产生,导致自身免疫和组织损伤[6]。最近的研究发现,IL-17A能够调节多种细胞的凋亡过程[7]。凋亡作为清除受损和有害细胞的程序性死亡过程,其与炎症反应密切相关。凋亡能够促进炎症病灶受损细胞的清除,而炎症反应过程中产生的炎症因子能够促进凋亡的增加[8-9]。然而,炎症反应和凋亡在氟诱导肝损伤中的相互作用关系,及IL-17A在该过程中的调节作用尚不清楚。
为此,本研究建立IL-17A基因敲除氟中毒小鼠模型,运用HE染色、ELISA、流式细胞术、免疫组化等技术检测肝的组织形态变化、炎症细胞含量、炎症因子水平和凋亡情况,进而探究氟对肝炎症反应和凋亡的影响,并明确IL-17A在该过程中的调节作用。
1 材料与方法
1.1 试验材料
C57小鼠和IL-17A基因敲除小鼠购自南模生物有限公司;氟化钠购自Sigma公司;HE染色试剂盒和BCA蛋白检测试剂盒购自索莱宝科技有限公司;ELISA试剂盒购自西唐生物科技有限公司;JC-1和Annexin V购自碧云天生物技术研究所;流式细胞抗体购自BD Pharmingen公司;免疫组化一抗和二抗购自武汉三鹰生物科技有限公司。
1.2 动物模型
24只野生型8周龄C57成年雄性小鼠随机分为对照组和NaF组,12只8周龄IL-17A基因敲除雄性小鼠作为KO+NaF组(体重20~25 g)。对照组饮用去离子水,NaF组和KO+NaF组饮用50 mg·L-1NaF的去离子水[5]。标准实验室条件下按照试验分组饲养12周。饲养结束后眼球采血,断颈处死小鼠,收集肝样品。
1.3 HE染色
小鼠处死后立即取适量大小肝组织固定于4%多聚甲醛。固定后的组织块进行冲洗、脱水、透明、浸蜡、包埋、切片制成5 μm的石蜡切片。然后,石蜡切片经过脱蜡至水、苏木精(5 min)和伊红(1 min)浸染、脱水、透明、封片。最后400倍镜下随机挑选10个区域观察组织中的异常病理变化并记录,并根据记录综合评定每组肝的损伤情况。
肝的组织形态。
1.4 透射电镜
透射电镜用于观察肝的超微结构变化。快速切取2 mm3的新鲜肝组织放入预冷的2.5%戊二醛溶液中固定2 h。组织块经乙醇和丙酮常规脱水,环氧树脂包埋。然后,用超薄切片机制作50~70 nm切片,并用醋酸铀染色液 (30 min)和柠檬酸铅染色液(10 min)染色。最后,用透射电镜观察肝超微结构。
1.5 ELISA检测
用预冷的PBS洗涤肝,并切取适量组织加入到10倍体积的PBS中。组织匀浆后,4 ℃ 离心(12 000g,15 min),吸取上清备用。然后,BCA测定蛋白浓度,并按照ELISA试剂盒说明书步骤检测匀浆液中炎症因子(IL-6、TNF-α、IL-17、TGF-β、INF-γ、IL-1β、IL-23)的含量。最后,计算每mg肝组织匀浆液中炎症因子含量=炎症因子含量/蛋白浓度。
1.6 流式细胞检测
本研究分别用CD3+/CD4+、CD3+/CD8+、CD68、CD11c、CD56、CC1、γδTCR流式抗体标记CD4+T细胞、CD8+T细胞、巨噬细胞、树突状细胞、自然杀伤细胞、肥大细胞、γδT 细胞。小鼠处死后,取新鲜肝组织制备细胞悬液。细胞悬液经过滤和洗涤后,用裂解液破碎红细胞。随后用流式抗体(1∶100)孵育30 min(对于胞内蛋白在抗体孵育前进行固定和破膜)。洗涤3次后上机(BD LSRFortessa细胞分析仪),并使用FlowJo处理数据。
1.7 免疫组化
石蜡切片常规脱蜡至水,3% H2O2消除内源性过氧化物酶,并用柠檬酸钠-EDTA抗原修复液100 ℃修复15 min。PBS洗涤后,兔抗鼠一抗(Caspase3和Cyt-c,1∶100)4 ℃孵育过夜,生物素化的山羊抗兔二抗(1∶1 500)室温下孵育1 h。然后,用DAB显色(试验过程中防止组织干燥),并用中性树脂封片。最后,用普通显微镜观察和图像分析软件(Image-Pro Plus)分析光密度值。
1.8 数据统计
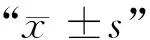
2 结 果
2.1 IL-17A基因敲除对氟诱导的肝组织形态和超微结构损伤的影响
通过HE染色和透射电镜观察组织病理学和超微结构变化(图1)。
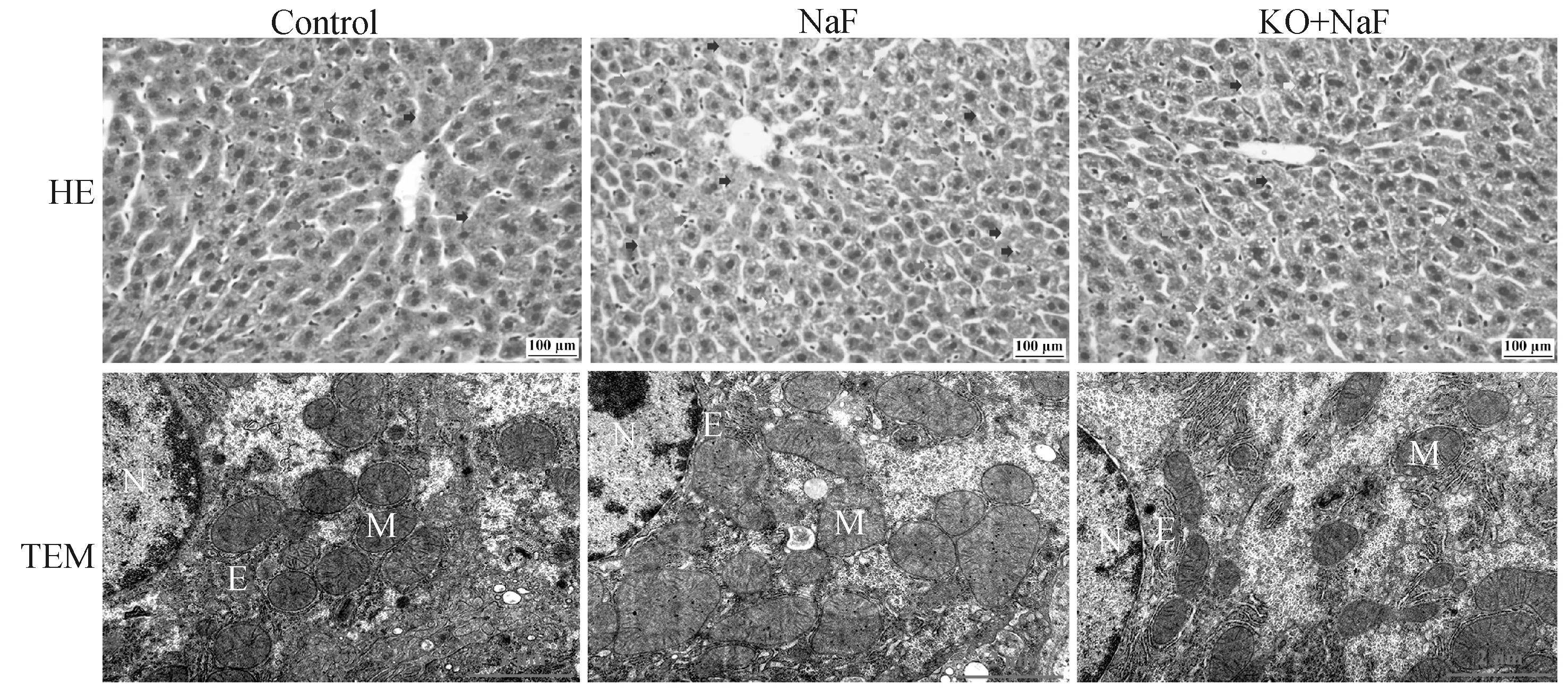
黄色箭头:炎症细胞;蓝色剪头:颗粒变性;黄色箭头:空炮变性;橙色箭头:核固缩;紫色箭头:核溶解;N. 细胞核;M. 线粒体;E. 内质网Yellow arrow: inflammatory cells; Blue arrow: particle denaturation; Yellow arrow: vacuolar denaturation; Orange arrow: nuclear solidification; Purple arrow: karyolysis; N. Nuclear; M. Mitochondria; E. Endoplasmic reticulum图1 HE染色(400×,n=6)和透射电镜结果(20 000×,n=3)Fig.1 The results of HE staining (400×, n=6) and transmission electron microscopy (TEM, 20 000×, n=3)
结果显示,对照组小鼠的肝形态结构正常,肝索排列紧密且有规则,线粒体和内质网形态正常,无明显异常。然而,氟暴露后肝中炎症细胞浸润增加,肝细胞间隙增大,颗粒变性、空泡变性、核固缩、核溶解肝细胞数量增多,线粒体畸形和脊损伤增加,内质网扩张和破碎增加。与NaF组相比,IL-17A基因敲除后肝组织结构损伤减轻,核固缩和核溶解细胞数量减少,炎症细胞浸润降低,畸形线粒体数量降低,内质网损伤减轻、数量增多。结果表明,IL-17A缓解了氟诱导的肝组织形态和超微结构损伤。
2.2 IL-17A基因敲除对氟诱导肝炎症因子表达改变的影响
本研究运用ELISA检测炎症反应过程中关键炎症因子(IL-6、TNF-α、IL-17、TGF-β、INF-γ、IL-1β、IL-23)在肝中的含量(图2)。与对照组相比,NaF组肝中TNF-α、IL-17、INF-γ、IL-23、TGF-β的含量显着升高(P<0.05,P<0.01),IL-1β的含量显着降低(P<0.01),然而,与NaF组相比,KO+NaF组肝中TNF-α、IL-17、INF-γ、IL-23、TGF-β的含量显着降低(P<0.05,P<0.01),IL-1β的含量显着升高(P<0.01)。IL-6含量在NaF组和KO+NaF组无显着变化。这些结果表明,IL-17A基因敲除缓解了氟诱导的肝炎症因子表达改变。
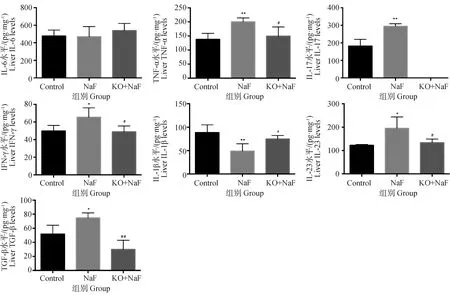
与对照组相比,*.P<0.05,**.P<0.01;#.P<0.05,与NaF组相比,##.P<0.01;下同*.P<0.05, **.P<0.01, vs control group; #.P<0.05, ##.P<0.01, vs NaF group; The same as below图2 肝炎症因子ELISA检测结果Fig.2 ELISA detection results of hepatic inflammatory factors (n=8,
2.3 IL-17A基因敲除对氟诱导肝CD4+T细胞和CD8+T细胞水平改变的影响
本研究用流式细胞术检测了肝中CD4+T细胞和CD8+T细胞。如图3所示,氟暴露后肝CD4+T细胞和CD4+T细胞/CD8+T细胞比值显着降低(P<0.05)。然而,与NaF组相比,IL-17A基因敲除显着增加了肝中CD4+T细胞和CD4+T细胞/CD8+T细胞比值(P<0.05)。CD8+T细胞在NaF组和KO+NaF组中无显着差异。
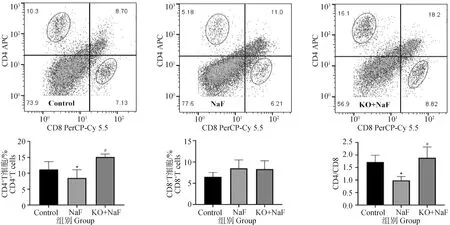
图3 CD4+T细胞和CD8+T细胞含量流式检测结果Fig.3 The flow cytometry results of CD4+ T cell and CD8+ T cell contents (n=6,
2.4 IL-17A基因敲除对氟诱导肝炎症细胞水平改变的影响
本研究通过流式细胞术检测肝中树突状细胞、巨噬细胞、肥大细胞、自然杀伤细胞、γδT 细胞含量观察IL-17A基因敲除对氟诱导肝炎症细胞水平改变的影响(图4)。结果显示,与对照组相比,NaF组肝中自然杀伤细胞和γδT 细胞显着降低,M2型巨噬细胞和树突状细胞显着升高(P<0.05)。IL-17A基因敲除后肝中树突状细胞与NaF组相比显着降低(P<0.05)。同时,KO+NaF组中的γδT 细胞和M2型巨噬细胞与对照组比无显着差异。上述结果表明,IL-17A基因敲除缓解了氟诱导的肝炎症细胞水平改变。
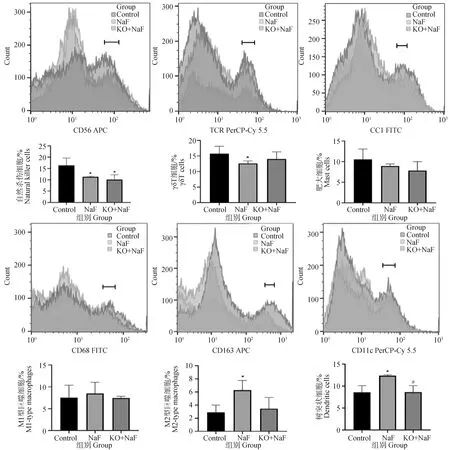
图4 巨噬细胞、树突状细胞、自然杀伤细胞、肥大细胞、γδT 细胞含量流式检测结果Fig.4 The flow cytometry results of macrophages, dendritic cells, natural killer cells, mast cells, and γδT cells contents (n=6,
2.5 IL-17A基因敲除对氟诱导肝细胞凋亡的影响
为探究IL-17A基因敲除对氟诱导的肝细胞凋亡的影响,本研究运用流式细胞术和免疫组化检测了凋亡肝细胞数和凋亡标志蛋白Caspase3和Cyt-c的蛋白表达水平。Annexin V和PI双染法是流式细胞术检测细胞凋亡的经典方法,流式结果如图5所示,与对照组相比,NaF组中肝细胞凋亡率显着增加(P<0.05)。然而,与NaF组相比,KO+NaF组中肝细胞凋亡率显着降低(P<0.05)。与流式结果相似,免疫组化结果显示(图6,表1),与对照组相比,NaF组肝中Caspase3和Cyt-c的蛋白表达水平极显着增加(P<0.01),而IL-17A基因敲除显着降低了Caspase3和Cyt-c蛋白表达与NaF组相比(P<0.05,P<0.01)。结果表明,IL-17A基因敲除减轻了氟诱导的肝细胞凋亡。

表1 肝Caspase3和Cyt-c蛋光密度值统计结果Table 1 The optical density value statistical results of Caspase3 and Cyt-c protein in the liver (n=5,
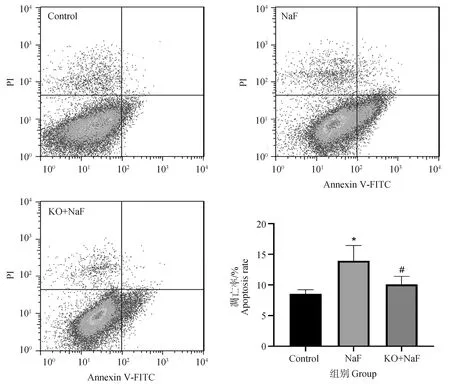
图5 肝细胞凋亡流式检测结果Fig.5 The flow cytometry results of hepatocyte apoptosis (n=6,
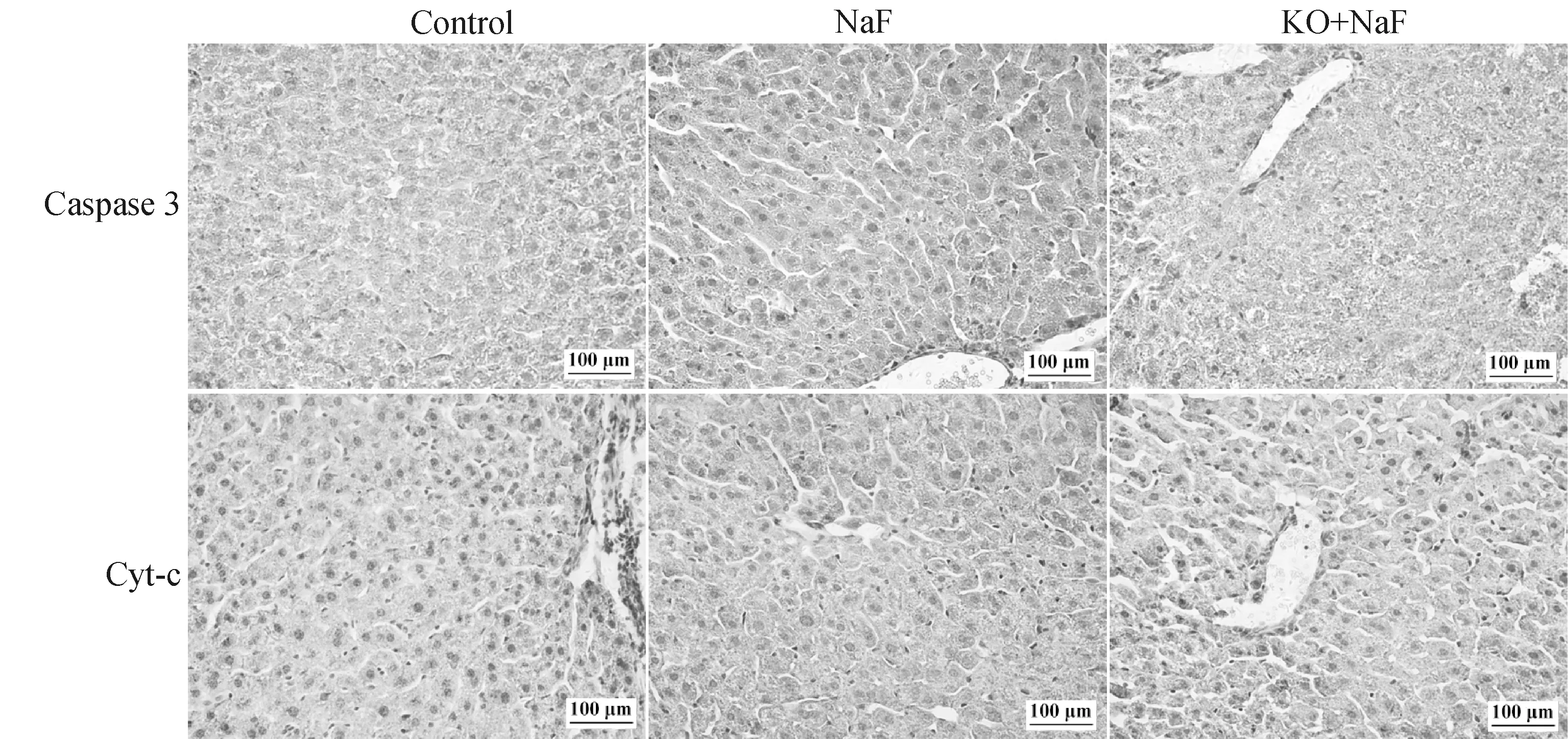
图6 肝Caspase3和Cyt-c蛋白免疫组化结果(400×)Fig.6 Caspase3 and Cyt-c immunohistochemical results (400×)
3 讨 论
氟中毒是一种广泛分布于世界许多国家和地区的人畜共患性地方病,中国是受其危害较为严重的国家之一[10]。氟中毒主要是由于长期慢性的氟暴露引起,并造成机体多种组织和器官的损伤。肝是氟中毒的重要靶器官,氟能够损伤肝的结构和功能,进而影响机体的物质代谢和有害物质的蓄积[1]。在一项关于美国青少年血氟含量与肝功能相关性调查的研究发现血清氟含量与肝功能呈负相关,氟暴露影响肝功能,反过来肝功能损伤会影响氟的吸收和代谢过程,这样的恶性循环进一步加重了氟对肝的危害[11]。同时,大量研究发现氟暴露能够导致肝中核固缩、核溶解、空泡变性、脂肪变性增多,线粒体和内质网损伤,以及炎症细胞的浸润[5,12]。与上述研究结果相似,在本研究中氟暴露导致了肝组织形态和超微结构损伤。在前期研究中,作者发现氟暴露后肝中IL-17A含量和炎症细胞浸润显着增加[5]。IL-17A作为重要的促炎因子,其在炎症的发生和发展过程中发挥重要的调节作用[13]。因此,作者推测IL-17A在氟诱导了肝的炎症损伤中发挥重要的作用。
IL-17A参与急慢性炎症过程,IL-17A的高表达能够促进免疫细胞的慢性浸润和多种炎症因子的表达,并最终导致炎症反应和组织损伤[14-15]。Zhang等[16]研究发现,IL-17A功能的阻断能够有效缓解胆汁淤积诱导的肝细胞坏死和肝损伤。同时,Gomes等[17]的研究也发现阻断IL-17A信号传导可减少脂肪变性和肝损伤,并预防肝细胞癌。相似地,在本研究中IL-17A的基因敲除缓解了氟诱导的肝组织形态损伤。为探究IL-17A基因敲除缓解肝损伤的机制,本研究进一步检测了肝中炎症因子和炎症细胞的变化情况。
炎症反应是机体抵御不良刺激的一种防御反应,但过强或长期的炎症反应能够诱导机体组织器官的功能损伤[18]。已有大量的研究报道外源性毒物能够导致肝中炎症因子和炎性细胞的水平改变,进而引发肝炎症[19]。同时,研究发现氟作为广泛存在与水和食物中的外源性毒物能够增加肝中炎症因子IL-2、IL-4、IL-6、IL-13、IL-21、TNF-α和TGF-β水平升高,以及IFN-γ/IL-4和IL-2/IL-10比值的降低[20]。与上述结果相似,本研究中氟暴露增加了肝中TNF-α、IL-17A、INF-γ、IL-23、TGF-β含量,以及M2型巨噬细胞和树突状细胞水平。同时,氟暴露降低了IL-1β、自然杀伤细胞、γδT 细胞和CD4+T细胞水平,以及CD4+T细胞/CD8+T细胞比值。IL-23、IL-17、TNF-α、INF-γ、IL-1β是关键的促炎细胞因子,TGF-β是关键的抑炎细胞因子[21]。巨噬细胞、树突状细胞、T淋巴细胞、自然杀伤细胞、γδT 细胞能够产生炎症因子,并介导炎症反应过程[18]。CD4+T细胞是机体免疫力的重要指标,其含量降低表明机体免疫力减弱。CD4+T细胞/CD8+T细胞比值是免疫调节的一项重要指标,其比值的异常表明免疫功能的紊乱[22]。这些结果表明氟暴露降低了肝的免疫能力,扰乱了肝炎症平衡,增加了肝的炎症反应。IL-17A作为炎症反应过程中重要的促炎因子,其不仅能够增加炎症因子的分泌,而且能够促进炎症细胞在受损组织中的浸润[14]。Zhang等[16]发现阻断IL-17A的作用显着减少肝中促炎细胞因子、中性粒细胞、巨噬细胞的流入。在本试验中,与NaF组相比,IL-17A基因敲除降低了肝中TNFα、IL-17A、INF-γ、IL-23、TGF-β的表达水平和树突状细胞含量,增加了IL-1β的表达水平。虽γδT 细胞和M2型巨噬细胞与氟组无显着差异,但其与对照组比无显着差异。有趣的是在本研究中IL-1β、自然杀伤细胞和γδT细胞在氟组降低,且IL-17A基因敲除增加了IL-1β水平。IL-1β结果可能的机制:IL-1β作为组织的细胞防御和组织修复的关键因子,其在细胞增殖、分化和凋亡等生物过程中发挥重要作用。肝作为自我修复能力较强的器官,受到氟的损伤后启动修复机制,抑制了IL-1β的表达。IL-17A基因的敲除能够缓解氟诱导的肝损伤,从而促进IL-1β的表达趋于正常。自然杀伤细胞和γδT细胞结果可能的机制:自然杀伤细胞和γδT细胞较为敏感,容易受到外源性毒物氟的刺激,激活细胞的凋亡程序,进而导致肝中自然杀伤细胞和γδT细胞数量的降低。自然杀伤细胞和γδT细胞作为机体重要的免疫细胞,其数量的减少能够降低机体的免疫力,进一步影响机体的炎症反应。同时,IL-17A基因的敲除未参与到上述诱导自然杀伤细胞和γδT细胞凋亡的过程。因此,IL-17A基因的敲除未缓解氟诱导的上述改变。在未来的研究中作者将进一步探究IL-1β、自然杀伤细胞和γδT细胞在氟致肝损伤中的作用机制。上述结果表明,IL-17A基因敲除缓解了氟诱导的肝炎症损伤。已有大量研究发现,炎症反应与细胞凋亡密切相关,一方面大量的炎症因子能够诱导细胞凋亡的发生,另一方面细胞凋亡能够清除炎症病灶内中性粒细胞和其他滞留细胞,限制组织损伤,促进炎症吸收[23]。因此,本研究进一步探究了IL-17A基因敲除对氟诱导的肝细胞凋亡的影响。
细胞凋亡是一种维持细胞微环境稳态的细胞自主性和程序性死亡方式。在生理条件下,机体通过凋亡去除不需要的和异常的细胞。而在病理条件下,受不利条件刺激细胞凋亡增加,导致正常细胞损伤[24]。已有研究发现,氟可导致肝细胞的凋亡增加[25-26]。例如,Ouyang等[25]报道氟通过Cyt-c/Caspase3/9通路诱导了鸭肝细胞的凋亡。Lu等[26]报道氟通过氧化应激和凋亡导致了小鼠肝损伤。与上述研究结果相似,本研究中氟暴露增加了肝中的凋亡细胞,以及凋亡关键基因Cyt-c和Caspase3的蛋白表达水平。Cyt-c是凋亡Caspase级联反应的关键蛋白酶激活因子,Caspase3是介导凋亡信号和执行细胞凋亡的关键激酶[8]。同时,本研究发现IL-17A基因敲除减轻了氟诱导的肝细胞凋亡,降低了Cyt-c和Caspase3的蛋白表达水平。在外源性毒物诱导肝炎症的研究发现,IL-17A作为重要炎症因子,其能够刺激多种细胞释放金属蛋白酶和炎症因子,促进炎症细胞的募集和肝炎症反应[14]。最近研究发现,IL-17A能够通过IL-17A受体通路调节细胞自噬、线粒体损伤、凋亡等生物过程[27-28]。例如,Kim等[27]研究发现IL-17A通过损伤线粒体功能诱导滑膜成纤维细胞的凋亡。因此,IL-17A缓解氟诱导的肝细胞凋亡的机制可能是氟引起了肝的炎症反应,炎症因子的大量释放刺激肝细胞的凋亡,同时氟作为外源性毒物能够激活IL-17A受体通路诱导肝细胞凋亡。然而,IL-17A基因的敲除缓解了氟诱导的肝炎症反应,减轻了炎症因子刺激的凋亡,并阻断了IL-17A受体通路诱导的肝细胞凋亡。
4 结 论
氟暴露损伤了肝的组织形态,并改变了肝内炎症因子(TNF-α、IL-17、INF-γ、IL-23、TGF-β、IL-1β)和炎症细胞(M2型巨噬细胞、树突状细胞、CD4+T细胞、自然杀伤细胞、γδT 细胞)的水平。同时,氟暴露诱导了肝细胞凋亡。然而,IL-17A基因敲除能够缓解氟诱导的肝炎症反应,并减轻氟诱导的肝细胞凋亡。本研究明确了IL-17A在氟诱导肝炎症反应和凋亡中的作用,并进一步揭示了氟肝毒性的内在机理,为氟中毒的防治提供了理论依据。



