王红丹,冯战启,娄桂予△,刘红彦,黄飞飞,刘石岭
(1.郑州大学人民医院/河南省人民医院医学遗传研究所,郑州 450003;2.河南省郑州市第一人民医院泌尿外科 450004)
·经验交流·
应用微阵列比较基因组杂交技术产前检测室间隔缺损胎儿2p16.3微缺失的临床研究*
王红丹1,冯战启2,娄桂予1△,刘红彦1,黄飞飞1,刘石岭1
(1.郑州大学人民医院/河南省人民医院医学遗传研究所,郑州 450003;2.河南省郑州市第一人民医院泌尿外科 450004)
目的 了解心脏室间隔缺损胎儿基因组拷贝数变化,寻找其致病的遗传学基础,探讨微阵列基因组杂交技术在产前诊断中应用的可行性。方法 用G显带核型分析技术分析胎儿及其父母外周血染色体,用微阵列比较基因组杂交技术(Array-CGH)进行患儿及其父母DNA拷贝数变异分析,经数据库比对和文献分析明确异常缺失区域的病理意义。结果 患儿父母外周血染色体核型分析未见异常;Array-CGH检测患儿发现染色体2p16.3区存在640×103bp的缺失,15q11区存在2 028×103bp的缺失,20p13区存在535×103bp的重复。结论 经查数据库及相关文献发现染色体2p16.3区域微缺失可能与胎儿室间隔缺损相关。
产前诊断;核酸杂交;基因组;可行性研究;微阵列比较基因组杂交;室间隔缺损
微阵列比较基因组杂交技术(Array-CGH)具有高分辨率、高通量、高灵敏性和高准确性。本研究应用Array-CGH对1例室间隔缺损的先天性缺陷胎儿羊水细胞进行高分辨率全基因组扫描分析,旨在寻找胎儿基因组中可能存在的病理性拷贝数变异(copynumbervariations,CNVs),丰富对室间隔缺损的病因学认识,探讨微阵列比较基因组杂交在产前分子细胞遗传学诊断中应用的可行性,为患儿父母再生育选择合适的产前诊断技术提供依据。
1 资料与方法
1.1 一般资料 孕妇,汉族,31岁。晚孕,因超声检查发现胎儿室间隔缺损至河南省人民医院医学遗传研究所行产前遗传咨询。患者与其配偶体检正常,非近亲结婚,无遗传家族史。为明确病因,经患者知情同意,进行了羊水穿刺术并抽取胎儿父母外周血进行细胞和分子遗传学检测。此项研究经过河南省人民医院医学伦理委员会审查批准。
1.2 方法
1.2.1 细胞遗传学核型分析 抽取胎儿父母静脉血各3mL,均肝素抗凝处理,取1mL接种于RPMI1640淋巴细胞培养液中进行培养,置于恒温箱37 ℃培养72h,收获前1h加入秋水仙素。常规细胞收获,制片,Giemsa染色显带,选择分散良好和显带清晰的核型镜下进行计数分析。
1.2.2DNA提取 取500μL血液,采用全血DNA柱式提取试剂盒(天根生化科技有限公司,北京)按照说明书操作进行。超微量分光光度计(NanoDrop2000,美国)测定DNA浓度和纯度,所提DNA样本A260/A280为1.80~1.90。
1.2.3Array-CGH检测分析 对患儿及其父母应用HumangenomeCGHMicroarray8×60K芯片(美国Agilent公司)进行检测,采用MicroarrayScanner(美国Agilent公司)及其他配套软件对芯片进行扫描和初步分析,检索UCSC、DECIPHER、ISCA等数据库对芯片实验结果进行进一步的及对比致病性鉴定。
1.2.4CNVs的判断和评价 基因组CNVs分析采用AgilentSurePrintG3HumanCGHMicroarrayKit,该芯片包含约6万个拷贝数标记探针。研究表明,致病的CNVs99.34%片段大于300×103bp[1],临床应用中建议针对200×103bp以上CNVs进行检测[2]。本检测仅针对染色体非整倍体性改变及200×103bp以上的片段缺失与重复,不排除有更小的染色体结构或基因片段异常的可能性。根据CNVs的性质不同可将其分为良性CNVs、可疑致病性CNVs及临床意义不明确的CNVs(variantsofuncertainsignificance,VOUS)。数据分析参考公开数据库UCSC、DECIPHER、ISCA等。
2 结 果
2.1 细胞遗传学检测结果 胎儿及其父母染色体核型正常。
2.2Array-CGH检测结果 胎儿Array-CGH检测发现2个微小缺失和1个微小重复,缺失发生在2p16.3区(hg19:52185415-52825075)和15q11区(hg19:20481702-22509254),大小分别为640×103bp和2 028×103bp,检测核型图见图1;重复发生在20p13区(hg19:439387-974841),大小约535×103bp。未见其他染色体存在探针拷贝数变化。其父母Array-CGH检测未发现拷贝数变化。
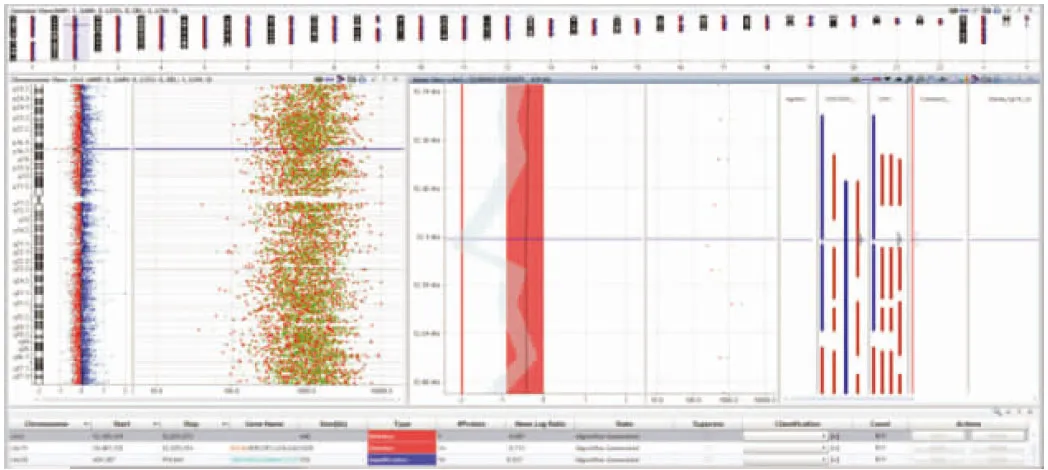
图1 Array-CGH检测核型图
3 讨 论
Array-CGH技术通过一次杂交试验就可以对全基因组DNACNVs进行检测,又被称为“分子核型分析”,具有高分辨率、高敏感性和高准确性等优点,对于检测染色体组微小缺失、重复等不平衡性重排具有独特优势,明显提高了基因组不平衡的检测率。2010年,国际细胞基因组芯片标准协作组(ISCAConsortium)推荐将Array-CGH作为对原因不明的发育迟缓、智力低下、多种体征畸形及自闭症患者的首选临床一线检测方法[1]。
染色体异常是先天发育畸形、发育迟缓和其他多种先天异常的主要原因。其中较小染色体不平衡畸变也可以引起智力低下、器官畸形和生长发育迟缓。而常规G显带技术对这些较小染色体不平衡畸变的检出率仅15%~40%[2]。目前,国外研究已经证实Array-CGH在针对智力低下、多发性先天畸形等患者的遗传诊断中的检出率为7%~11%[3-4]。染色体微缺失或重复引起的综合征常见的有Williams综合征、DiGeorge或velo-eardio-facial综合征及Prader-Willi综合征等,其引起的伤残程度及其给家庭、社会造成的负担严重[5]。而导致这些综合征的染色体异常片段通常比较细小,因此往往不能被常规方法识别。
先天性心脏病(congenitalheartdefect,CHD)是最常见的先天缺陷,发生率为活产儿的1%和自然流产的10%[6],其中染色体异常占其发生原因的8%,环境因素占2%,余为多因子因素引起[7]。报道最多的引起CHD的微缺失发生在22q11基因群,发生率1.4~2.5%[8-10]。许争峰等[11]对163例的CHD进行研究发现7.36%CHD存在22q11关键区微缺失。另据报道,与胎儿心脏正常发育密切相关的还有5q35区段、心脏特异性同源盒基因及Jagged1基因等[12]。
本例Array-CGH检测胎儿发现染色体2p16.3区域存在640×103bp的缺失(hg19:52185415-52825075),15q11区域存在2 028×103bp的缺失(hg19:20481702-22509254),20p13区域存在535×103bp的重复(hg19:439387-974841)。经查阅公开数据库UCSC、DECIPHER、ISCA等未见染色体15q11区域2 028×103bp缺失和20p13区域535×103bp重复的致病性报道,且在健康人群中存在这两种CNVs。而数据库显示2p16.3区域缺失与智力障碍、癫痫、语言发育障碍、面部发育畸形等多种临床特征相关,但未见该区域与室间隔缺损相关性的报道,本例发现该缺失区段也与室间隔缺损相关,丰富了数据库信息。关于该区域致病的文献报道也很多,Meng等[13]的研究显示该区域的突变可能与原发性青光眼相关,Zhen等[14]的研究显示该区域可能与生育相关,另有研究显示该区域的微缺失与发育迟缓、身材矮小和先天性膈疝等相关[15]。
总之,应用Array-CGH,在一个室间隔缺损的胎儿基因组中检出了一个未报道过的染色体2p16.3新发缺失,明确诊断了本病例2p缺失的断裂位点、片段大小乃至基因序列,也为临床表型与基因型关系的研究提供了较为完整的信息。2p16.3区域缺失具有多种临床特征,临床特征的可辨识性不高,如果明确诊断仍需要分子细胞遗传学实验分析,该缺失与疾病表型的相关性还有待于进一步研究去澄清。
[1]MillerDT,AdamMP,AradhyaS,etal.Consensusstatement:chromosomalmicroarrayisafirst-tierclinicaldiagnostictestforindividualswithdevelopmentaldisabilitiesorcongenitalanomalies[J].AmJHumGenet,2010,86(5):749-764.
[2]葛运生,孔辉,曾寰,等.高分辨微阵列比较基因组杂交技术在临床复杂染色体异常遗传诊断中的应用[J].中华检验医学杂志,2013,36(1):46-49.
[3]ParkSJ,JungEH,RyuRS,etal.Clinicalimplementationofwhole-genomearrayCGHasafirst-tiertestin5 080preandpostnatalcases[J].MolCytogenet,2011(4):12.
[4]PickeringDL,EudyJD,OlneyAH,etal.Array-basedcomparativegenomichybridizationanalysisof1 176consecutiveclinicalgeneticsinvestigations[J].GenetMed,2008,10(4):262-266.
[5]廖灿,符芳,杨昕,等.应用微阵列比较基因组杂交技术产前诊断DiGeorge综合征一例[J].中华医学遗传学杂志,2011,28(2):238.
[6]TriedmanJK,NewburgerJW.Trendsincongenitalheartdisease:thenextdecade[J].Circulation,2016,133(25):2716-2733.
[7]TongYF.MutationsofNKX2.5andGATA4genesinthedevelopmentofcongenitalheartdisease[J].Gene,2016,588(1):86-94.
[8]DeGregoriM,CicconeR,MaginiP,etal.Crypticdeletionsareacommonfindingin"balanced"reciprocalandcomplexchromosomerearrangements:astudyof59patients[J].JMedGenet,2007,44(12):750-62.
[9]AgergaardP,OlesenC,ØstergaardJR,etal.Theprevalenceofchromosome22q11.2deletionsin2,478childrenwithcardiovascularmalformations.Apopulation-basedstudy[J].AmJMedGenetA,2012,158(3):498-508.
[10]孙碧君,吴冰冰,郭晓红,等.22q11.2微缺失综合征20例临床表型分析[J].中华实用儿科临床杂志,2015,30(8):589-592.
[11]许争峰,易龙,莫绪明,等.先天性心脏病患者22q11微缺失检测及相关分析[J].中华医学遗传学杂志,2006,23(3):250-255.
[12]廖晓波,周新民,杨一峰,等.应用基因微阵列技术筛选法乐四联症相关基因[J].中国循环杂志,2008,23(2):147-150.
[13]MengN,MaJ,XiaL,etal.AssociationbetweenSNPs(rs1533428,rs12994401,rs10202118)onchromosome2p16.3andprimaryopenangleglaucoma[J].CurrEyeRes,2015,40(8):839-846.
[14]ZhenXM,SunYM,QiaoJ,etal.Genome-widecopynumberscaninChinesepatientswithprematureovarianfailure.[J]BeijingDaXueXueBao,2013,45(6):841-847.
[15]Bermudez-WagnerK,JengLJ,SlavotinekAM,etal.2p16.3microdeletionwithpartialdeletionoftheneurexin-1geneinafemalewithdevelopmentaldelays,shortstature,andacongenitaldiaphragmatichernia[J].ClinDysmorphol,2013,22(1):22-24.
国家自然科学基金资助项目(81501336,81270488)。 作者简介:王红丹(1983-),助理研究员,硕士,主要从事生殖医学方面研究。△
E-mail:louguiyu@qq.com。
10.3969/j.issn.1671-8348.2016.23.030
R
B
1671-8348(2016)23-3256-02
2016-04-02
2016-06-30)



