耿娜娜,吴明松,,郑 翔,杨 蕾,王宏阳,李学英△
(1.遵义医学院口腔学院,贵州遵义 563000;2.贵州省高等学校口腔疾病研究特色重点实验室/遵义医学院医学与生物学研究中心,贵州遵义 563000;3.遵义医学院医学遗传学教研室,贵州遵义 563000)
有研究显示,2015年中国肝癌男女新发病分别为343 700例和122 300例,其中男性死亡310 600例,女性死亡111 500例,占癌症相关死因的17.2 %和11.1 %[1]。原发性肝癌(primary liver cancer,PLC)在我国发病率较高,其中肝细胞癌(hepatic carcinoma,HCC)占PLC的90%以上[2-3]。顺铂(cis-dichlorodiamine platinum,CDDP)是治疗HCC的主要药物之一,但其单药有效率比较低[3],毒副作用也较大。研究表明,单宁酸(tannic acid,TA)可与其他药物协同发挥抗癌作用,如与丝裂霉素C、5-氟尿嘧啶联用抗胆管癌[4],与三氧化二砷(As2O3)联用抗人类白血病[5],与CDDP联用抗卵巢癌[6]等。此外,TA能够抑制人肝癌HepG2细胞的生长增殖[7-9],但其分子机制尚不清楚。有研究表明口服TA较为安全,并被作为食品添加物[6]。因此,TA可能作为一种低毒的化疗药物增敏药物,参与癌症的预防和治疗。
内质网主要负责蛋白质的合成、修饰与转运、信号转导等,当这些过程受到干扰时,会导致蛋白质的折叠、翻译后修饰、基因表达等发生紊乱,从而引起内质网应激(endoplasmic reticulum stress,ERS)[10]。ERS介导的细胞凋亡与众多癌症的发生、发展等有关联[11-12]。研究表明,CDDP能够通过激活ERS促进肝癌HepG2细胞的凋亡[13]。TA是否能够协同增强CDDP抗肝癌,并通过ERS途径诱导肝癌细胞凋亡,尚少见报道。本研究旨在探讨TA协同增强CDDP抗肝癌HepG2细胞的作用,并通过体外实验探索ERS凋亡通路在该协同效应中的作用。
1 材料与方法
1.1材料 人肝癌细胞株HepG2,来源于中国科学院细胞库。TA(C76H52O46,含量大于或等于95%)购自Sigma公司,CDDP注射液[Cl2(NH3)2Pt]购自山东齐鲁制药公司,RPMI-1640培养基购自Gibco公司,胎牛血清(FBS)购自浙江天杭生物科技有限公司,噻唑蓝(MTT)购自BBI公司,4′6-二脒基-2-苯基吲哚(DAPI)购自Biosharp公司,M-MLV逆转录酶购自Promega公司,dNTP Mix(10 mmol/L)购自上海生工生物公司,RNAiso TM Plus购自TaKaRa Biotechnology公司,PCR引物(表1)由上海生工生物公司合成,Sso Fast Eva Green supermix实时荧光定量PCR(q-RT-PCR)试剂盒购自Bio-Rad公司,鼠抗人葡萄糖调节蛋白(glucose regulated protein,GRP)94单抗、兔抗人GRP78多抗和兔抗人β-actin单抗、辣根过氧化物酶标记的多抗均购自Protein-Tech Group公司。酶标仪Model 680、PCR仪CFX Connect TM Optics Module均购自Bio-Rad公司,倒置荧光显微镜IX73购自Olympus公司,超微量分光光度计Nanodrop 2000购自Thermo Scientific公司,GOLD-SIM二氧化碳培养箱购自西盟国际公司。
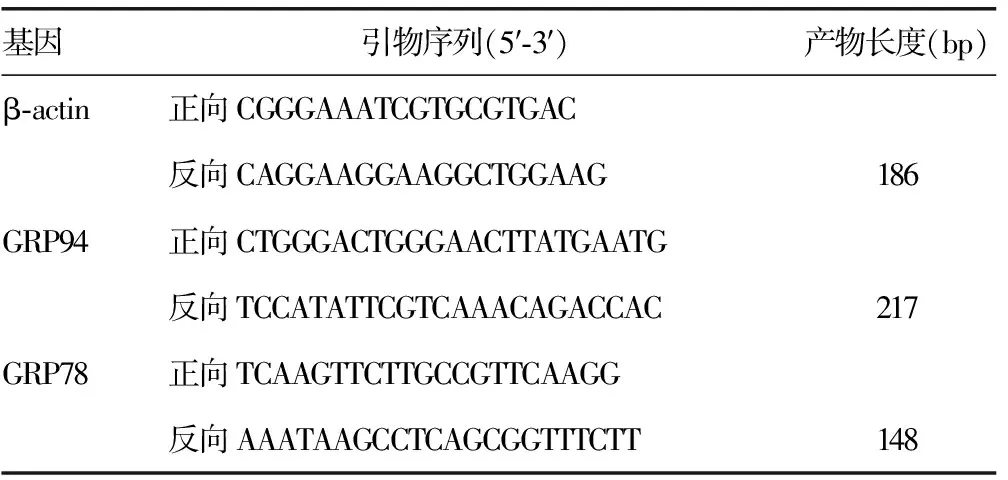
表1 PCR引物序列及扩增长度
1.2方法
1.2.1细胞培养 将人肝癌细胞系HepG2培养于含10% FBS、2.0 g/L NaHCO3、100 μg/mL链霉素、100 U/mL青霉素的RPMI-1640培养基中,于37 ℃、5%CO2饱和湿度的培养箱中培养,每2~3 天传代1次。
1.2.2细胞的生长抑制作用 采用MTT法检测并计算细胞的存活率[14]。取对数生长期HepG2细胞接种于96孔培养板中,每孔1×104个。静置培养24 h后,分别加TA 0(对照组)、90.00、180.00、270.00、360.00、450.00、540.00 μmol/L和CDDP 0(对照组)、0.60、1.20、1.80、2.40、3.00、3.60 μg/mL,每组设5个复孔。继续培养24 h后,每孔加入10.00 μL浓度为5.00 mg/mL 的MTT溶液;再培养4 h后,弃上清液,每孔中加入100.00 μL二甲基亚砜,轻轻振荡10 min。用酶标仪测定570 nm波长下各样品的吸光度(A)值,各复孔的A取平均值,计算细胞的存活率,其中空白对照组细胞存活率记为100%。用等效线图解法观察TA和CDDP的协同作用,其中等效线图中的斜线为两种药物联合产生100%最大抑制效应时的相加等效线,当二者的剂量比例点位于等效线上时,表现为相加作用;位于等效线下方,表现为协同作用;位于等效线上方,表现为拮抗作用。药物的协同作用还可通过药物联合作用指数(combination index,CI)和药物剂量降低指数(dose reduction index,DRI)来判断[15]。以(D)1和(D)2表示两种药物联用达到某一特定生长抑制率时的使用浓度;(DX)1和(DX)2表示达到与联合用药相同的生长抑制率时,两种药物单独使用的浓度。CI<1表明药物具有协同作用;CI=1表明药物具有相加作用;CI>1表明药物具有拮抗作用。DRI表示在给定的抑制率下,与药物单独使用相比较,药物联合使用达到同一抑制率时,药物剂量所降低的倍数。计算公式如下:
细胞存活率(%)=(实验组A值-阴性对照组A值)/(空白对照组A值-阴性对照组A值)
(1)
CI=(D)1/(DX)1+(D)2/(DX)2
(2)
(DRI)1=(DX)1/(D)1
(3)
(DRI)2=(DX)2/(D)2
(4)
1.2.3细胞形态与细胞核变化的观察 DAPI是一种能够穿透细胞膜与核DNA结合的荧光染料,可用于检测细胞核的形态变化和细胞凋亡,正常的细胞核较为完整,染色质均匀;而凋亡细胞的染色质发生固缩,边缘化,严重时发生细胞核裂解,产生核碎片等。取对数期HepG2细胞接种于6孔板,每孔3×105个(接种约3.00 mL)。药物处理24 h后,经丙酮固定,DAPI染色后,于显微镜下观察细胞和细胞核的形态,并照相。
1.2.4q-RT-PCR检测 药物处理24 h后,收集细胞,提取细胞总RNA,根据试剂盒方法,逆转录合成cDNA。q-RT-PCR反应体系体积为10.00 μL,包括cDNA 1.00 μL、Sso Fast Eva Green supermix 5.00 μL,上游引物0.50 μL,下游引物0.50 μL,无菌水3.00 μL。PCR反应程序:94 ℃预变性60 s;95 ℃变性20 s,56 ℃(GRP78和β-actin)或63.5 ℃(GRP94)退火30 s,40个循环周期。每个样品设置3管重复。以β-actin作为内参,采用2-△△CT的方法,用3次重复的平均值计算基因相对表达量。
1.2.5Western blot检测 药物处理24 h后,提取细胞总蛋白质,采用BCA法测定蛋白质的浓度,取一定体积量蛋白样品,进行变性、电泳、转膜、封闭、孵育一抗(GRP78 1∶2 000;GRP94 1∶2 000;β-actin 1∶10 000)和二抗(1∶2 000)、曝光与显影,晾干。
2 结 果
2.1TA与CDDP协同抑制肝癌细胞生长 TA和CDDP均能明显抑制HepG2细胞的存活率,且均呈剂量性依赖(P<0.01),TA和CDDP的半数抑制率(IC50)浓度分别为360.00 μmol/L(TA组,图1A)和1.80 μg/mL(CDDP组,图1B)。采用两种药物IC50一半浓度的方法联合处理HepG2细胞,TA、CDDP组对HepG2的抑制率分别为(19.10±6.00)%、(17.50±4.00)%,而TA 360.00 μmol/L+CDDP 1.80 μg/mL(联合组)抑制率为(60.30±2.00)%,明显高于TA、 CDDP组(P<0.01),见图1C。等效线图中TA与CDDP的剂量比例点绝大多数位于等效线下方(图1D)。不同剂量的TA与CDDP联用产生的抑制率为9.00%~70.00%(IC9~IC70)时,其CI分别为0.44、0.48、0.70、0.75、1.35,大多数均小于1;不同比例的TA与CDDP联用的DRI分为1.40~4.00和1.60~5.30,见表2。
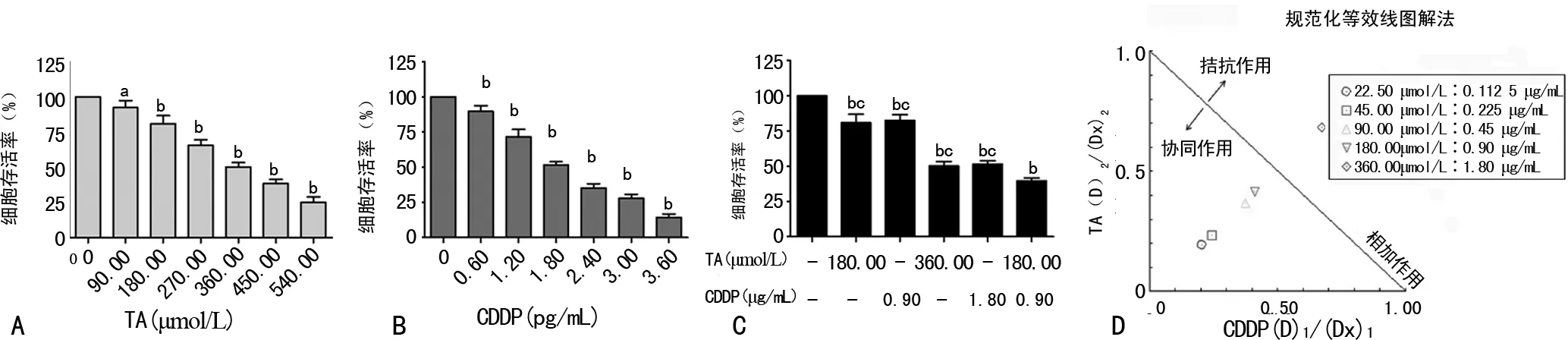
A:单纯TA;B:单纯CDDP;C:二者联用;D:规范化等效线图解法;a:P<0.05,b:P<0.01,与对照组(0)比较;c:P<0.01,与联合组比较
图1不同剂量TA与CDDP对肝癌HepG2细胞生长的抑制作用
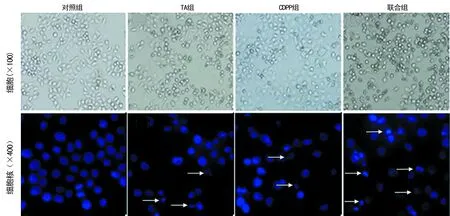
图2 各组肝癌HepG2细胞的形态及细胞核变化
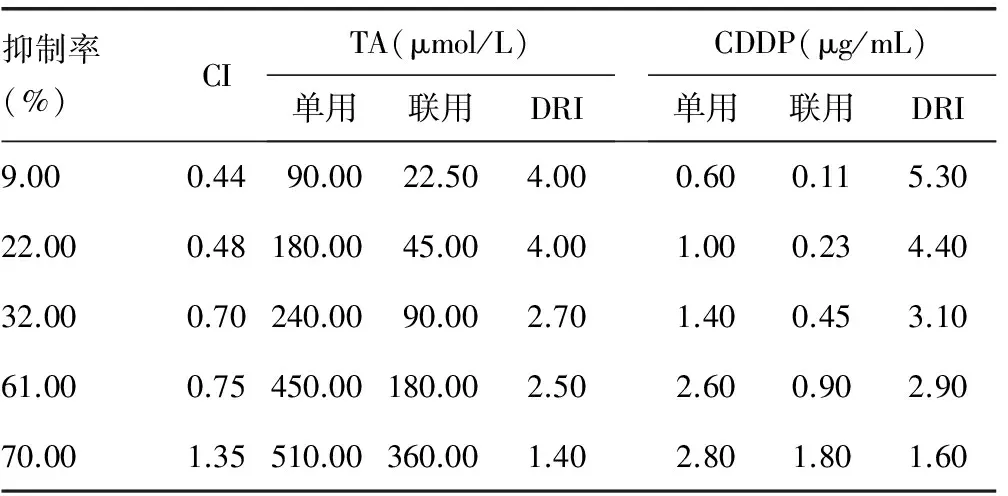
表2 TA与CDDP的CI与DRI情况
2.2TA增加CDDP的促肝癌细胞凋亡作用 凋亡细胞的基本形态变化主要有:细胞体变小、变圆,连接消失,与周围的细胞脱离,细胞质浓缩,折光度降低等。药物处理24 h后,对照组细胞贴壁良好,呈梭形或多角形,轮廓较为清晰,折光度均一;TA、CDDP组部分细胞开始脱壁,细胞体积皱缩、变圆,折光度降低;联合组出现大量脱壁死亡细胞和细胞碎片,折光度明显降低。药物处理24 h后DAPI染色显示,对照组细胞核较为完整,为椭圆形或圆形,染色质较为均匀,呈淡蓝色;TA组、CP组部分细胞核固缩,出现大小不同的核裂解碎片(凋亡小体);而联合组多数细胞核固缩,细胞染色质聚集,部分细胞核边缘呈现不规则,染色较深,呈现蓝色荧光,出现更多的凋亡小体,表现出典型的细胞凋亡形态学变化,见图2。
2.3TA与CDDP协同上调肝癌细胞ERS水平 药物处理24 h后,联合组细胞GRP78、GRP94 mRNA及蛋白的表达量明显高于TA、CDDP组(P<0.05),见表3、图3。
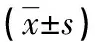
表3 各组GRP78、GRP94 mRNA及蛋白水平比较
a:P<0.01,与对照组比较;b:P<0.01,c:P<0.05,与联合组比较

图3 各组肝癌HepG2细胞GRP78和GRP94蛋白表达
3 讨 论
本研究发现TA与CDDP单独用药均能够抑制HepG2细胞的生长增殖,且均呈剂量性依赖;当使用二者IC50的一半浓度进行联合用药后,上述效应加剧,抑制率明显高于各单药组,表明TA与CDDP具有协同抗HepG2的作用。等效线图解法、CI、DRI,细胞及细胞核的形态学观察结果进一步证明了二者的协同作用。已有研究表明,药物产生的毒副作用主要与用药剂量密切相关,因此通过降低药物使用剂量能够降低其毒副作用[16],TA与CDDP 的协同抑制HepG2细胞的作用,能够降低CDDP的使用剂量,减少其毒副作用,并且TA本身毒性较低,可作为CDDP的增敏药物,参与癌症的预防和治疗,作者认为TA具有潜在的临床应用价值。
当环境因素极大地扰乱内质网动态平衡的过程时,内质网将进行应激,并激活各种ERS反应。一方面,内质网会启动各种调节机制以减轻这些损伤,使细胞适应环境的胁迫,以促进细胞的存活;另一方面,如果细胞无法适应环境的胁迫,长时间的ERS将会引起细胞凋亡的发生[17-18]。GRP78和GRP94是内质网蛋白的分子伴侣,被认为是ERS的标志性分子。GRP78,即相对分子质量为78×103调节血糖的蛋白,也被称为ERS传感器结合免疫球蛋白(the immunoglobulin heavy chain binding protein,BiP)。GRP94是另一种主要的GRP,相对分子质量为94×103。在非ERS状态下,GRP78与蛋白激酶RNA样内质网激酶(PERK)、肌醇酶1α(IRE1α)、激活转录因子6(ATF6)形成稳定的配合物,并保持无活性状态[19]。GRP78通过阻止PERK和IRE1α二聚体的形成,来抑制其活性[19];与ATF6结合阻断高尔基体定位信号,使ATF6稳定结合在内质网膜,阻止ATF6的进一步激活[20]。在ERS状态下,内质网中积累的错误折叠蛋白与GRP78蛋白竞争,使GRP78从PERK、IRE1α和ATF6 上解离,从而失去其抑制作用,引起下游相关信号转导[20]。
ERS介导的细胞凋亡与众多癌症的发生、发展等有关。癌细胞自身创造的低氧、低pH值和低营养等不利的微环境都有助于引起ERS。但癌细胞能够通过多种方式去适应这种环境应激,以阻止ERS诱导的细胞凋亡相关信号途径[18]。当癌细胞无法适应环境的胁迫,持续的ERS将会诱导癌细胞凋亡的发生,如MESE等[21]研究发现GRP78表达上调能够使人表皮细胞癌A431细胞株对CDDP的敏感性增强。王涛等[22]研究发现GRP94表达上调能够增加肺癌细胞株NCI-H460对CDDP的敏感性。本研究显示,用药24 h后,TA、CDDP组与联合组GRP78和GRP94表达均有所上调,且联合组上调最高,表明ERS是TA和CDDP协同促进HepG2细胞凋亡的靶点之一。其机制可能是TA通过阻断蛋白质从内质网到高尔基体之间的运输,诱导HepG2细胞的ERS 水平升高,高水平的ERS通过激活PERK-ATF4、IRE1α-XBP1和ATF6信号通路来抑制细胞DNA修复酶的活性,从而协同性增加CDDP对癌细胞的DNA 损伤作用[23-25],最后激活Caspase 蛋白家族级联反应[26],诱导细胞凋亡的发生。然而,关于ERS 的PERK-ATF4、IRE1α-XBP1和ATF6 信号通路激活的具体分子机制有待进一步研究。
综上所述,TA和CDDP能够协同增强肝癌HepG2细胞的凋亡,其协同作用与ERS途径的激活有关,为TA与CDDP应用于临床治疗肝癌提供了理论基础。
[1]CHEN W Q,ZHENG R S,BAADE P D,et al.Cancer statistics in China,2015[J].CA Cancer J Clin,2016,66(2):115-132.
[2]中国抗癌协会肝癌专业委员会,中华医学会肝病学分会肝癌学组,中国抗癌协会病理专业委员会,等.原发性肝癌规范化病理诊断指南(2015年版)[J].解放军医学杂志,2015,40(11):865-872.
[3]中华人民共和国卫生部.原发性肝癌诊疗规范(2011年版)[J].临床肝胆病杂志,2011,27(11):1141-1159.
[4]NAUS P J,HENSON R,BLEEKER G,et al.Tannic acid synergizes the cytotoxicity of chemotherapeutic drugs in human cholangiocarcinoma by modulating drug efflux pathways[J].J Hepatol,2007,46(2):222-229.
[5]CHEN K S,HSIAO Y C,KUO D Y,et al.Tannic acid-induced apoptosis and -enhanced sensitivity to Arsenic trioxide in human leukemia HL-60 cells[J].Leuk Res,2009,33(2):297-307.
[6]SUN Y Y,ZHANG T H,WANG B D,et al.Tannic acid,an inhibitor of poly(ADP-ribose) glycohydrolase,sensitizes ovarian carcinoma cells to cisplatin[J].Anticancer Drugs,2012,23(9):979-990.
[7]宛春雷,金哲雄.仙鹤草鞣质成分的抗肿瘤作用研究[J].黑龙江医药,2011,24(1):29-31.
[8]陈小会,周云凯,蒋福升,等.棕榈子提取物抗肿瘤活性研究[J].海峡药学,2012,24(6):265-267.
[9]杨琳,李善俊.MTT法检测蕲艾鞣酸体外抗肿瘤的活性[J].中国民康医学,2013,25(8):1-3.
[10]STUTZMANN G E,MATTSON M P.Endoplasmic reticulum Ca2+handling in excitable cells in health and disease[J].Pharmacol Rev,2011,63(3):700-727.
[11]ZHAO Y C,ZHU C L,LI X F,et al.Asterosaponin 1 induces endoplasmic reticulum stress-associated apoptosis in A549 human lung cancer cells[J].Oncol Rep,2011,26(4):919-924.
[12]SHI Y H,DING Z B,ZHOU J,et al.Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis[J].Autophagy,2011,7(10):1159-1172.
[13]PARK I J,KIM M J,PARK O J,et al.Cryptotanshinone induces ER stress-mediated apoptosis in HepG2 and MCF7 cells[J].Apoptosis,2012,17(3):248-257.
[14]THAVAMANI B S,MATHEW M,DHANABAL S P.In vitro cytotoxic activity of menispermaceae plants against HeLa cell line[J].Anc Sci Life,2013,33(2):81-84.
[15]LI C J,CHU C Y,HUANG L H,et al.Synergistic anticancer activity of triptolide combined with cisplatin enhances apoptosis in gastric cancer in vitro and in vivo[J].Cancer Lett,2012,319(2):203-213.
[16]李大魁,张石革,张继春.药学综合知识与技能[M].北京:中国医药科技出版社,2011:301-309.
[17]MEIR O,DVASH E,WERMAN A,et al.C/EBP-beta Regulates Endoplasmic Reticulum Stress-Triggered Cell Death in Mouse and Human Models[J].PLoS One,2010,5(3):e9516.
[18]MARTIONON F.Targeting endoplasmic reticulum signaling pathways in cancer[J].Acta Oncol(Madr),2012,51(7):822-830.
[19]HETZ C.The unfolded protein response:controlling cell fate decisions under ER stress and beyond[J].Nat Rev Mol Cell Biol,2012,13(2):89-102.
[20]SHEN J,CHEN X I,HENDERSHOT L,et al.ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals[J].Dev Cell,2002,3(1):99-111.
[21]MESE H,SASAKI A,NAKAYAMA S,et al.Analysis of cellular sensitization with cisplatin-induced apoptosis by glucose-starved stress in cisplatin-sensitive and -resistant A431 cell line[J].Anticancer Res,2001,21(2A):1029-1033.
[22]王涛,王琪,裴复阳.GRP94表达在大细胞肺癌NCI-H460细胞对顺铂耐药中的作用[J].大连医科大学学报,2011,33(6):517-520.
[23]NAGELLERKE A,BUSSINK J,VAN DER KOGEL A J,et al.The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response[J].Radiother Oncol,2013,108(3):415-421.
[24]STROME E D,WU X W,KIMMEL M,et al.Heterozygous screen in Saccharomyces cerevisiae identifies dosage-sensitive genes that affect chromosome stability[J].Genetics,2008,178(3):1193-1207.
[25]DUFEY E,URRA H,HETZ C.ER proteostasis addiction in cancer biology:Novel concepts[J].Semin Cancer Biol,2015,15(33):40-47.
[26]黄亨平,吴明松.内质网应激--肿瘤治疗的新靶点[J].现代医药卫生,2014,30(17):2600-2603.



