胡朝恩,钟大鹏,艾智华△
(1.四川省攀枝花市中心医院重症医学科 617000;2.成都军区总医院内分泌科,成都 610083)
糖尿病发病率在全球范围内持续高速增长,已严重影响人类健康和生活质量;糖尿病是胰岛素相对或绝对缺乏引起碳水化合物、脂肪和蛋白质代谢紊乱,以血浆葡萄糖升高为主要表现的代谢性疾病,也有学者称之为“糖脂病”[1]。Toll样受体(TLRs)是最重要的一类模式识别受体,可选择性识别病原相关分子模式,激活天然免疫系统,构成抵御病原微生物入侵的第一道屏障。有研究发现,Toll受体2(TLR2)的激活能够介导上调白细胞介素-1β(IL-1β)、白细胞介素-6(IL-6)的表达,诱导胰岛炎症,导致胰岛素抵抗[2];TLR4水平的升高参与调节胰岛β细胞功能障碍[3];而抑制或基因敲除小鼠体内的TLR2和Toll受体4(TLR4)受体能预防2型糖尿病发生胰岛素抵抗,减弱糖尿病和早期糖尿病肾病的轻度炎症状态。主要表达于胰腺组织细胞内的Toll受体3(TLR3),能识别组织细胞损伤、凋亡释放的内源性产物[4]。主要通过Toll/IL-1受体接头蛋白分子(TRIF)途径激活信号分子核转录因子κB(NF-κB)、干扰素调节因子-3(IRF-3)等,引起局部细胞凋亡和促进炎症[5]。本研究旨在探讨高糖高脂对胰岛β细胞的损伤及机制,观察TLR3、肿瘤坏死因子-α(TNF-α)、IL-1β、IL-6、CRP、补体C3及补体C4的表达,现报道如下。
1 材料与方法
1.1材料与试剂
1.1.1试验细胞 小鼠胰岛β细胞株(NIT-1)购于上海拜力生物科技有限公司。
1.1.2试剂 细胞培养试剂购自Gibeo公司,CCK-8 检测试剂盒购自武汉博士德生物工程有限公司;胰岛素放免试剂盒购自北京北方生物技术研究所;棕榈酸(PA)购自美国Sigma 公司;总RNA提取试剂盒、反转录聚合酶链反应(RT-PCR)试剂盒购自大连宝生物工程有限公司;IL-1β、IL-6、TNF-α、CRP,补体C3、C4购自美国eBioscience公司;其他常用试剂均为国产分析纯产品。
1.2方法
1.2.1高糖、高脂培养基制备和细胞模型建立
1.2.1.1高糖、高脂培养基制备 (1)高脂培养基制备:高脂培养基中含棕榈酸0.25 mmol/L,具体方法:取10 mL 0.1 mol/L的NaOH溶液溶解51.3 mg棕榈酸配制成20 mmol/L的棕榈酸母液;然后取1.67 mL 30%胎牛血清清蛋白与1.25 mL 20 mmol/L的棕榈酸母液充分混匀,最后再加入含1 mL双抗溶液95 mL于1640培养基中,充分混匀;(2)高糖培养基制备:高糖培养基中含葡萄糖25 mmol/L,具体方法为100 mL高糖培养基即取90 mL DMEM高糖培养基、10 mL胎牛血清、1 mL双抗混合。
1.2.1.2细胞模型建立 NIT-1细胞待生长至60%~70%融合后,弃原培养基,分别加入高糖、高脂培养基诱导48 h后建立高糖、高脂胰岛β细胞模型。
1.2.2细胞培养、传代和分组 小鼠NIT-1培养于含15%胎牛血清、100 U/mL青霉素和100 mg/mL链霉素的DMEM低糖培养基中,隔天换液。根据细胞密度按1∶(1~3)的比例传代。待细胞数量足够时取对数生长期细胞进行试验分组:正常组(C组)、高糖组[G组,含葡萄糖(Glucose) 25 mmol/L]、高脂组(Z组,含PA 0.25 mmol/L)、高糖高脂组(GZ组,含Glucose 25 mmol/L和PA 0.25 mmol/L)。
1.2.3细胞活力检测 取对数生长期密度约为80%~90%的NIT-1细胞,用适量稀释的0.25%胰蛋白酶消化为单细胞悬液,按每孔7×103个细胞密度接种于96孔板,置于37 ℃、5%CO2培养箱培养24 h,待细胞贴壁后,将细胞分为C组、G组、Z组和GZ组,分别给予完全培养基、高糖培养基、高脂培养基和高糖高脂培养基培养48 h。采用CCK-8试剂盒检测细胞活力;CCK-8试剂盒是基于WST-8[2-(2-甲氧基-4-硝苯基)-3-(4-硝苯基)-5-(2,4-二磺基苯)-2H-四唑单钠盐]广泛应用于细胞增殖和细胞毒性的一种快速高灵敏度检测试剂盒,水溶性四唑盐WST-8在电子载体1-Methoxy PMS作用下,可被细胞线粒体中的一些脱氢酶还原成水溶性橙黄色甲臜染料,颜色深浅与细胞增殖呈正比,与细胞毒性呈反比;细胞增殖越多越快,则颜色越深,而细胞毒性越大,则颜色越浅;对于同样的细胞,颜色深浅和细胞数目呈良好线性关系;使用酶标仪测定450 nm波长处的OD值,间接反映活细胞数量。
1.2.4TLR3 mRNA检测
1.2.4.1总RNA提取 (1)吸取培养液,使用预冷1×磷酸盐缓冲液(PBS )清洗2次;(2)按试剂盒推荐最佳剂量,向培养细胞中加入适量的裂解液Buffer RL(使用前已加入50×DTT Solution溶液),在水平位置轻轻摇晃,使裂解液均匀分布于细胞表面,以便充分裂解细胞,然后使用移液枪反复吹打细胞,使其脱落;(3)将内含细胞的裂解液转移至无菌离心管中,用移液枪反复吹吸直至裂解液中无明显沉淀,裂解液室温静置2 min;(4)将去gDNA吸附柱安放到2 mL的Collection Tube上,将裂解液转移到去gDNA吸附柱中,12 000 r/min离心1 min,弃去gDNA吸附柱,保留2 mL Tube 中的滤液;(5)加入与液体等体积的 70%乙醇,使用移液枪将溶液混合均匀,立即将混合液(含沉淀)全部转入到RNA Spin Column(含2 mL Collection Tube)中,12 000 r/min离心1 min,弃滤液;(6)将 RNA Spin Column 放回到 2 mL Collection Tube 中,将500 μL 的Buffer RWA 加入至RNA Spin Column中,12 000 r/min离心30 s,弃滤液;(7)将含等体积100%乙醇的600 μL Buffer RWB沿管壁四周加入至RNA Spin Column中,12 000 r/min离心30 s,弃滤液,再重复本操作1次;(8)将RNA Spin Column 重新安置于2 mL Collection Tube上,12 000 r/min 离心2 min,再将RNA Spin Column 安置于1.5 mL的RNase Free Collection Tube上,向RNA Spin Column 膜中央处加入50~200 μL的0.1% DEPC处理水,室温静置5 min;(9)12 000 r/min 离心2 min洗脱 RNA。
1.2.4.2紫外定量和检测 先用稀释用的TE溶液将分光光度计调零,然后取少量RNA溶液用TE稀释(1∶100)后,读取其在分光光度计260 nm和280 nm处的OD值,测定RNA溶液的浓度和纯度。
1.2.4.3相关引物合成 按照Genebank 给出的基因序列,设计小鼠TLR3 mRNA引物,引物系列:上游,5′-TCA CTT GCT CAT TCT CCC TT-3′,157 bp;下游,5′-GAC CTC TCC ATT CCT GGC-3′。
1.2.4.4RT-PCR 严格按照TaKaRa的One Step SYBR®PrimeScriptTMRT-PCR Kit(Perfect Real Time)DRR066A试剂盒说明书进行操作。
1.2.5IL-1β、IL-6、TNF-α、CRP及补体C3、C4检测 取对数生长期密度为80%~90%的NIT-1细胞,用适量稀释的0.25%胰蛋白酶消化为单细胞悬液,按每孔7×103个细胞密度接种于96孔板,置于37 ℃、5%CO2培养箱培养24 h,待细胞贴壁后,将细胞分为C组、G组、Z组和GZ组,分别给予完全培养基、高糖培养基、高脂培养基和高糖高脂培养基培养48 h,收集细胞上清液,分别检测TNF-α、IL-1β、IL-6、CRP及补体C3、C4的表达。本试验采用夹心法酶联免疫吸附试验(ELISA)测定各检测指标,具体操作严格按照ELISA说明书进行。
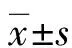
2 结 果
2.1高糖高脂抑制胰岛β细胞增殖 与C组比较,G组、Z组及GZ组胰岛β细胞增殖水平明显受到抑制,差异均有统计学意义(P<0.01),且胰岛β细胞增殖情况呈C组向G组、Z组、GZ组逐渐下降的趋势,G组胰岛β细胞增殖率低于C组,GZ组又低于G组和Z组,差异均有统计学意义(P<0.01),但G组与Z组胰岛β细胞增殖率差异无统计学意义(P>0.05)。可见糖脂毒性协同抑制胰岛β细胞增殖的能力远远大于单独的高糖或高脂作用(图1)。
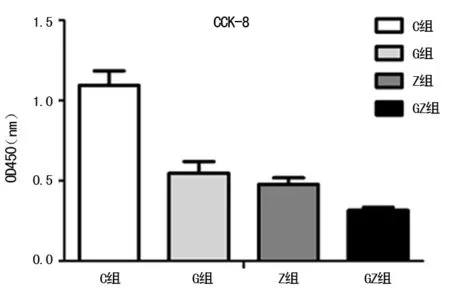
图1 各组胰岛β细胞活力比较(以OD值表示)
2.2高糖高脂诱导胰岛β细胞TLR3、TNF-α、IL-1β、IL-6、CRP、补体C3及补体C4过表达
2.2.1TLR3表达情况 与C组比较,各组胰岛β细胞TLR3 mRNA的表达明显增高,差异均有统计学意义(P<0.05);GZ组TLR3 mRNA的表达明显高于其他各组,差异均有统计学意义(P<0.05);而Z组与GZ组TLR3 mRNA表达差异无统计学意义(P<0.05)。糖脂协同作用产生的毒性作用明显高于高糖、高脂的单独作用(图2)。
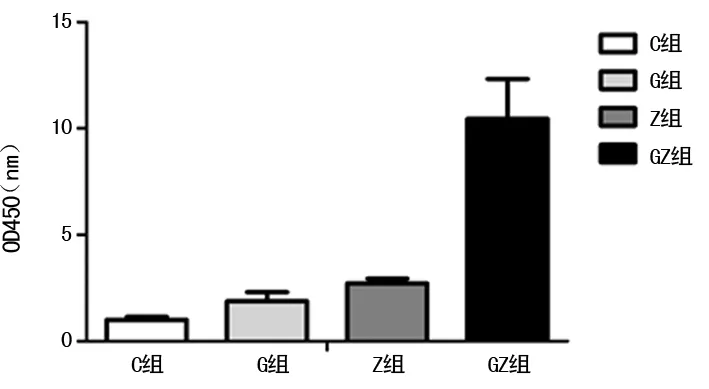
图2 各组TLR3 mRNA表达情况(以OD值表示)
2.2.2TNF-α、IL-1β、IL-6、CRP、补 体C3及补体C4表达情况 见表1。由表1可见,G组、Z组、GZ组TNF-α的表达明显高于C组,差异均有统计学意义(P<0.05),而G组、Z组、GZ组间TNF-α的表达差异无统计学意义(P>0.05);经高糖、高脂处理后,各组胰岛β细胞IL-1β和IL-6的表达均明显增加,与C组比较差异均有统计学意义(P<0.05);CRP在各组表达均明显高于C组,差异有统计学意义(P<0.05),且G组和GZ组CRP水平明显高于Z组,差异有统计学意义(P<0.05);经过高糖、高脂处理后,各组胰岛β细胞表达补体C3与C4水平均明显高于C组,差异均有统计学意义(P<0.05)。
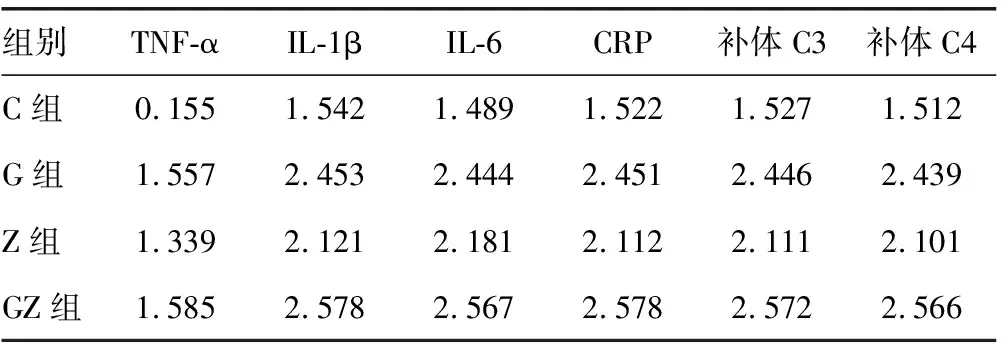
表1 各组TNF-α、IL-1β、IL-6、CRP、补体C3及补体C4表达情况(以OD值表示)
3 讨 论
糖尿病与免疫炎症的关系最早有学者于1993年在动物试验中发现。之后许多研究也证实,糖尿病患者血清中各种炎症标志物水平较对照者明显增加,如IL、CRP、TNF-α等[6-8];在糖耐量减低或受损阶段就存在炎症因子水平的变化,慢性低水平的持续性炎症可能是糖尿病持续发展的关键。“炎症与糖尿病”的关系已成为全球内分泌学者共同关注的热点。无论1型糖尿病还是2型糖尿病,都是一种机体系统性轻度炎症状态,而这种轻度炎症状态来源于内源性和外源性配体刺激先天免疫细胞产生和分泌前炎症因子[9]。目前普遍认为,糖尿病是由细胞因子介导的一种自身免疫和慢性低度炎性疾病,表现为一些非特异性的炎性标志物水平升高,包括免疫炎症细胞(如白细胞)、急性期反应蛋白(如CRP)、细胞因子(如TNF-α、IL系列)、脂肪因子、补体系统和细胞黏附分子等[10-11]。
有研究证实,炎症标志物参与了糖尿病及其并发症的发生和发展,但具体作用机制还尚待研究[12-13]。TNF-α、IL-6及CRP是目前公认的敏感的亚临床炎症标志物[14]。CRP是主要的急性时相蛋白,主要由TNF-α、IL-6等刺激肝脏产生,具有免疫识别和调节功能,能激活补体系统,进一步释放炎症介质,加剧免疫炎症,如此形成一个反馈循环[15]。炎症可减少周围组织对葡萄糖的利用,降低β细胞对葡萄糖的敏感性,诱导胰岛素抵抗,抑制胰岛β细胞生长和分化,诱导β细胞凋亡和功能障碍,最终导致胰岛素分泌相对或绝对不足。
本研究应用高糖、高脂刺激后发现,胰岛β细胞大量表达TLR3 mRNA、TNF-α、IL-1β、IL-6、CRP及补体C3、C4,诱导β细胞炎症,进而导致β细胞功能障碍、细胞增殖和胰岛素分泌受到明显抑制。高糖、高脂环境下TLR3过量表达,在受到内、外源性配体刺激后,激活TLR3信号通路,主要通过TRIF途径完成一系列信号转导,使NF-κB和IRF-3活化,最终导致多种炎症因子(TNF-α、IL-1β、IL-6等)合成和释放,TNF-α、IL-6、IL-1β又促进CRP 生成,而CRP能激活补体系统,诱导补体C3、C4增加,进一步释放炎症介质,加剧免疫炎症,如此形成一个反馈循环;抑制胰岛β细胞的生长活力和胰岛素分泌,诱导β细胞凋亡和功能障碍,最终导致糖尿病的发生和发展。
综上所述,糖尿病的发生和发展可能是糖脂代谢紊乱和慢性免疫炎症等多因素共同作用的结果,而且这些因素可能在血糖调节受损阶段就已出现。糖脂代谢紊乱与慢性低度炎症有极为密切的关系;长期高糖、高脂刺激会诱导胰岛β细胞产生大量炎症因子,引发β细胞的慢性免疫炎症,导致胰岛β细胞的损伤和功能障碍,抑制胰岛素分泌和β细胞增殖,造成胰岛素分泌相对或绝对缺乏;慢性免疫炎症又反过来加剧糖脂代谢进一步紊乱,最终二者共同促进糖尿病及其并发症的发生和发展。
[1]GUO H,INGOLIA N T,WEISSMAN J S,et al.Mammalian microRNAs predominantly act to decrease target mRNA levels[J].Nature,2010,466(738):835-840.
[2]WANG P,HOU J,LIN L,et al.Inducible microRNA-155 feedback promotes type Ⅰ IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1[J].J Immunol,2010,185(10):6226-6233.
[3]ZHOU H,HUANG X,CUI H,et al.miR-155 and its star-form partner miR-155* cooperatively regulate type Ⅰ interferon production by human plasmacytoid dendritic cells[J].Blood,2010,116(26):5885-5894.
[4]ANDROULIDAKI A,ILIOPOULOS D,ARRANZ A,et al.The kinase akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs[J].Immunity,2009,31(2):220-231.
[5]CURTALE G,CITARELLA F,CARISSIMI C,et al.An emerging player in the adaptive immune response:microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes[J].Blood,2010,115(2):265-273.
[6]CHASSIN C,KOCUR M,POTT J,et al.miR-146a mediates protective innate immune tolerance in the neonate intestine[J].Cell Host Microbe,2010,8(4):358-368.
[7]JURKIN J,SCHICHL Y M,KOEFFEL R,et al.miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation[J].J Immunol,2010,184(9):4955-4965.
[8]CEPPI M,PEREIRA P M,DUNAND-SAUTHIER I,et al.MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells[J].Proc Natl Acad Sci USA,2009,106(8):2735-2740.
[9]LACKEY D E,OLEFSKY J M.Regulation of metabolism by the innate immune system[J].Nat Rev Endocrinol,2016,12(1):15-28.
[10]RECCHIUTI A,KRISHNAMOORTHY S,FREDMAN G,et al.MicroRNAs in resolution of acute inflammation:identification of novel resolvin D1-miRNA circuits[J].FASEB J,2011,25(2):544-560.
[11]THULASINGAM S,MASSILAMANY C,GANGAPLARA A,et al.miR-27b*,an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells[J].Mol Cell Biochem,2011,352(1/2):181-188.
[12]LIU G,FRIGGERI A,YANG Y,et al.miR-147,a microRNA that is induced upon toll-like receptor stimulation,regulates murine macrophage inflammatory responses[J].Proc Natl Acad Sci U S A,2009,106(37):15819-15824.
[13]XU Z,XIAO S B,XU P,et al.miR-365,a novel negative regulator of interleukin-6 gene expression,is cooperatively regulated by Sp1 and NF-kappaB[J].J Biol Chem,2011,286(24):21401-21412.
[14]RODRIGUEZ A,VIGORITO E,CLARE S,et al.Requirement of Bic/microRNA-155 for normal immune-function[J].Science,2007,316(5824):608-611.
[15]TAGANOV K D,BOLDIN M P,CHANG K J,et al.NF-kappaB-dependent induction of microRNA miR-146,an inhibitor targeted to signaling proteins of innate immune responses[J].Proc Natl Acad Sci U S A,2006,103(33):12481-12486.



