王佳庆 赵志刚 梅升辉首都医科大学附属北京天坛医:药学部,北京100050
HPLC法测定人血浆中万古霉素浓度的不确定度评定
王佳庆赵志刚梅升辉
首都医科大学附属北京天坛医:药学部,北京100050
目的评定液相色谱(HPLC)法测定人血浆中万古霉素(VCM)浓度的不确定度。方法采用Ag1ient ZORBAX SB-C18色谱柱(4.6 mm×250 mm,5 μm),流动相为30 mmo1/L磷酸二氢钾缓冲溶液-乙腈(91.5:8.5 pH值3.38),流速1.1 mL/min,检测波长236 nm,柱温35℃。分析并评定HPLC法测定人血浆中VCM浓度过程中的不确定度来源并进行合成。结果人血浆中VCM低浓度(2.48 μg/mL)和高浓度(172.2 μg/mL)的扩展不确定度分别为0.168 μg/mL和9.64 μg/mL(P=95%,k=2)。结论用HPLC法测定人体血浆中万古霉素浓度的不确定度在高浓度时主要由基质效应、生物样品的配制及重复性引入,在低浓度时主要由回收率、基质效应及生物样品配制引入。
高效液相色谱法;万古霉素;血清;不确定度
[Abstract]Objective To eva1uate the uncertainty on the determination of vancomycin(VCM)in human p1asma by HPLC method.Methods The chromatography was performed on an Ag1ient ZORBAX SB-C18(4.6 mm×250 mm,5 μm)co1umn with the mobi1e phase consisting of 30 mmo1/L potassiumdihydrogen phosphate-acetonitri1e(91.5:8.5,pH 3.38)at the f1ow rate of 1.1 mL/min.UV detection was set at 236 nm and the co1umn temperature was set at 35℃.The uncertainty sources in the determinationof VCM were ana1yzed and the uncertainty was eva1uated and combined.Results The expanded uncertainty of VCM at 1ow(2.48 μg/mL)and high(172.2 μg/mL)concentrations were 0.168 μg/mL and 9.64 μg/mL respective1y(P=95%,k=2).Conclusion The uncertainty on the determination of VCM in human p1asma by HPLC method is main1y caused by recovery,matrix effectand samp1e preparation for 1ow concentrations,and the matrix effect, samp1e preparation and repeatabi1ity for high concentrations.
[Key words]HPLC method;Vancomycin;Human p1asma;Uncertainty
万古霉素(vancomycin,VCM)属于三环糖肽类抗菌药物,临床上主要用于敏感革兰阳性菌,特别是严重耐药的金黄色葡萄球菌和表皮葡萄球菌所致的感染,因为此药有耳、肾毒性和血栓性静脉炎等严重的不良反应,故临床应用时常进行血药浓度监测以调整给药方案[1]。万古霉素、去甲万古霉素的浓度测定方法国内外多有报道[2-5],本实验建立了用HPLC法测定血浆中VCM的浓度。本文采用了测量不确定度来评价分析测试结果的质量。当不确定度值越小时,分析测试结果与真实值越接近,其质量就越高,数据也越可靠[6]。本文根据相关规范和指南评价了HPLC法测定血清中VCM浓度的不确定度,为改进实验方法、提高检测准确度提供参考。
1 仪器与试剂
日本岛津C1ass-VP高效液相色谱系统;UPW-20n多效蒸馏水器;Mett1er AE 163电子天平;ARK PH5-25酸度计;TGL-20M型高速台式冷冻离心机(湘仪器械有限公司);旋片式真空泵(上海市精工真空设备厂,型号:2XZ-1)。
VCM对照品(SIGMA,批号:BCBD7944V);去甲万古霉素内标对照品(中国药品生物制品检定所;批号:30339-200303);乙腈:色谱纯(Fisher公司,批号:67-55-1);超纯水。
2 方法与结果
2.1色谱条件
以去甲万古霉素作为内标物质,按内标法定量分析。液相色谱柱(Agi1ent,ZORBAX SB-C18,184.6 mm× 250 mm,5 μm,批号:880975-902),A相流动相为30 mmo1/L磷酸二氢钾缓冲液(pH值3.38),B相流动相为乙腈,流速为1.1 mL/min,柱温35℃,在波长为236 nm处检测,91.5%的A相与8.5%的B相等度洗脱18 min。
2.2对照品和内标溶液的配制
精密称取VCM对照品21.38 mg,定容到5.0 mL得S8(4 mg/mL)。取2.5 mL S8定容到5 mL,得S7。取2.5 mL S7定容到5.0 mL得S6。取2.5 mL S6定容到5.0 mL得S5。取2.5 mL S5定容到5.0 mL得S4,取2.0 mL S4定容到5.0 mL得S3,取2.5 mL S3定容到5 mL得S2,取4.0 mL S2定容到5.0 mL得S1。S8~S1对照品溶液浓度依次为4000、2000、1000、500、250、100、50、20 μg/mL,高、中和低浓度质控样品的浓度分别为3600、500、50 μg/mL。
精密称取内标对照品5.15 mg,流动相定容到10 mL作为储备液,配制浓度为0.515 mg/mL的内标液CIS备用。
2.3对照品、质控品和待测样品的配制
万古霉素的系列对照品溶液和内标液CIS各10μL加入190 μL空白血浆中,涡旋30 s后加入60 μL高氯酸,涡旋30 s后13 000 r/min离心10 min,取上清液20 μL进样。
取待测样品的配制内标液CIS10 μL加入200 μL待测样品血浆中,涡旋30 s后加入60 μL高氯酸,涡旋30 s后13000 r/min离心10 min,取上清液20 μL进样。
2.4标准曲线
线性回归方程:y=ax+b,其中:x为万古霉素浓度,y为万古霉素与内标的峰面积之比,a为斜率,b为截距。
2.5数学模型的建立
万古霉素浓度的计算:C=(y-b)/a×CIS×f,其中:y为万古霉素与内标物的峰面积之比,CIS为内标物的浓度,f为温度对万古霉素和内标物浓度测定的影响。
2.6测量不确定度来源的分析
本实验建立的方法选择性好,基本无干扰,日内的精密度(RSD)低浓度<13.17%,高浓度<0.61%,日间的RSD低浓度<10.61%,高浓度<0.41%。回收率在(98.92±4.35)%和(100.07±4.22)%之间。方法在室温放置、冷冻放置和反复冻融3次的稳定性良好。本文建立了不确定度的测试方法来考察实验结果的准确性。通过分析测定过程可以得知测量不确定度的来源主要有:标准品的称量、重复性、仪器允差、工作液的配制和生物样品的制备、标准曲线拟合、基质效应及萃取过程、温度、样品不均匀性和VCM浓度等诸多因素。见图1。
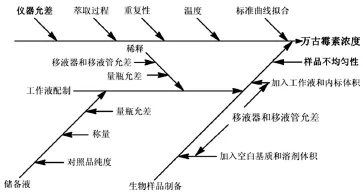
图1 测量不确定度来源分析
2.7测量不确定度的评定
2.7.1重复性(用A类评定程序)
重复性引入的不确定度,用质控样品重复测定值评估,低浓度(L)和高浓度(H)的质控样品共有3组(m=3),每组平行测量了5次(n=5),结果见表1。用贝塞尔公式计算合并样本偏差:
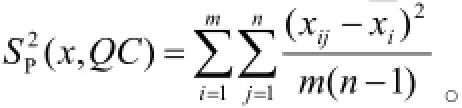
其中:i为组数(i=1,2,…m),j为每组平行测定次数(j=1,2,…n)。每组5个测定值平均值的标准偏差为:,质控的相对标准测量不确定度为:172.2 μg/mL,则低、高浓度的相对标准测量不确定度分别为:ur(1,L)=ur(x,L)=0.020,ur(1,H)=ur(x,H)= 0.0092。
表1 质控样品重复测定数据(±s,n=15)
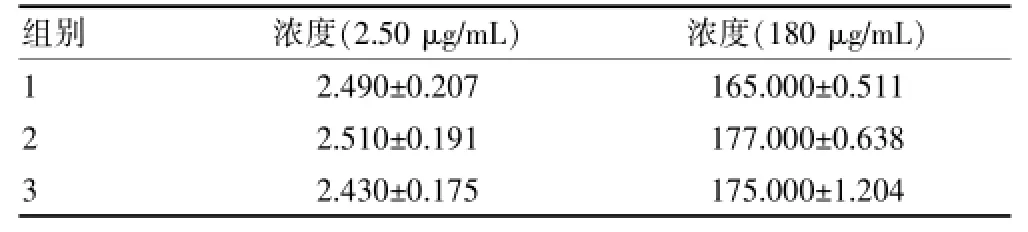
表1 质控样品重复测定数据(±s,n=15)
组别浓度(2.50 μg/mL)浓度(180 μg/mL)123 2.490±0.207 2.510±0.191 2.430±0.175 165.000±0.511 177.000±0.638 175.000±1.204
2.7.2对照品称量(用B类评定程序)
对照品称量引起的不确定度一般可以表示为:
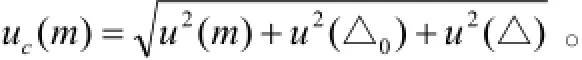
其中,天平的重复性误差u(m)在重复性实验中已经计算,此处不再重复计算。随机变量半宽a=0.5 e,天平检定分度值e=0.01 mg。减重称量时,自动调零作为一次扣皮,则a0=a,按均匀分布,包含因子k=,则自动调零u(△0)和天平的非线性误差u(△)引起的标准测量不确定度为:0.0029(mg)

天平的标准测量不确定度计算为:
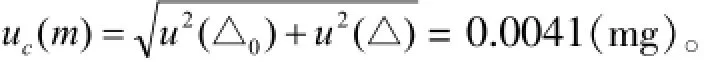
VCM称量的质量是21.38 mg,因为内标物的称量不会对VCM的浓度测定造成影响,所以对照品称量的相对标准测量不确定度计算为:
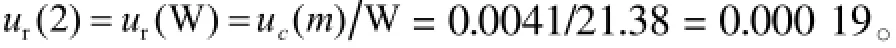
2.7.3对照品配制和生物样品制备(用B类评定程序)
2.7.3.1容量瓶引入的测量不确定度由JJG196-2006常用玻璃量器检定规程[13]查得A级5 mL容量瓶的容量允差为±0.010 mL。按三角形分布,包含因子k=,则由5 mL容量瓶引入的相对标准测量不确定度为
2.7.3.2移液器引入的测量不确定度实验使用的Eppendorf移液器规格有:2~20 μL(P1)和20~200 μL(P2)。P1和P2在吸取10、60、190 μL时的相对最大允差分别为±1.2%、±1.0%、±0.6%。按三角形分布,包含因子,移液器引入的相对标准测量的不确定度分别计算为:
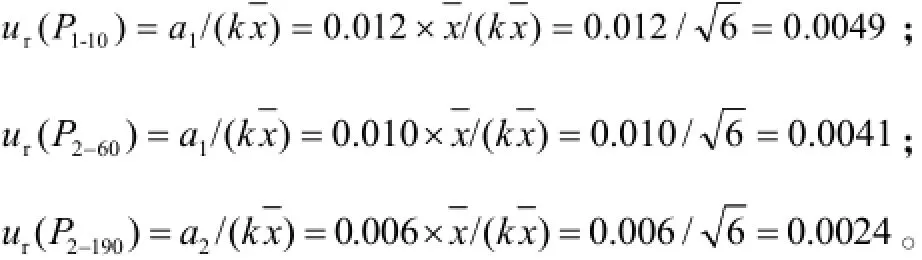
2.7.3.3移液管引入的测量不确定度实验所用移液管(P)为A级,规格为2 mL和5 mL,最大允差分别为± 0.012 mL和±0.025 mL。按三角形分布,包含因子,移液管引入的相对标准测量不确定度分别计算为:
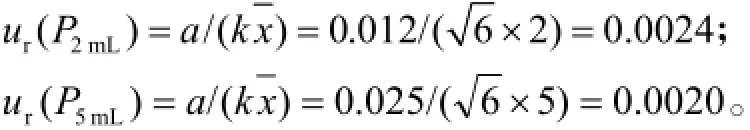
2.7.3.4对照品溶液配制过程中引入的测量不确定度对照品溶液配制过程中使用了2 mL移液管1次,5 mL的移液管6次,5 mL的容量瓶8次。内标物的配制不影响测定,因此对照品溶液配制引入的相对标准测量不确定度为:
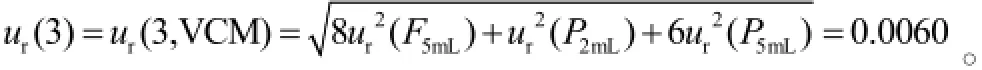
2.7.3.5含药的标准品和质控品配制引入的测量不确定度含药的标准品(S)和质控样品(QC)配制方法虽然相同,但是所用移液器的型号和次数分别为:P1吸取10 μL 2次,P2吸取60 μL 1次,190 μL 1次,则配制含药标准品和质控品的相对标准测量不确定度为:
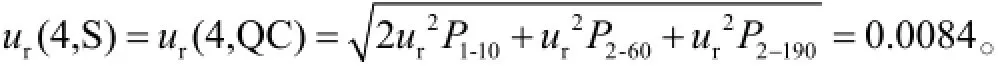
则样品制备引入的相对标准测量不确定度为:
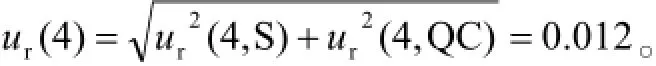
2.7.4萃取过程中引入的测量不确定度(用B类评定程序)
萃取过程(RE)引入的不确定度可由萃取回收率进行评估。评估方法:分别用空白溶剂和血浆配制高、低浓度质控样品,平行测定5次来评估回收率。计算公式:回收率(%)=血浆配制样品萃取后的峰面积/经过前处理的空白基质配制样品的峰面积×100%,内标归一化的回收率(%)=VCM的回收率/IS的回收率× 100%。低、高浓度质控样品内标归一化的回收率分别为(98.92±4.35)%和(100.07±4.22)%,则和回收率ur(5)引入的相对标准测量不确定度为:
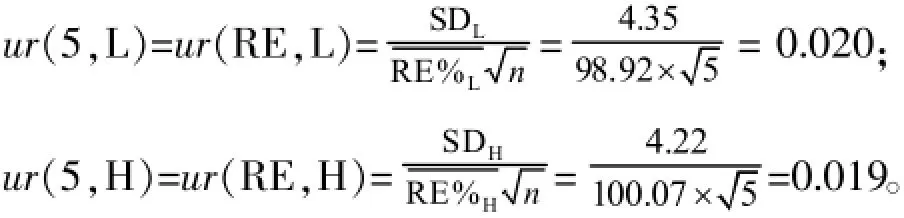
2.7.5仪器量化引入的测量不确定度(用B类评定程序)
根据岛津液相仪检定证书,定量测量重复性RSD=2%,按均匀分布原则,k=?,则仪器量化引入的相对标准测量不确定度可以表示为:
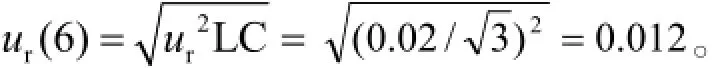
2.7.6线性拟合过程中引入的不确定度(用B类评定程序)
标准曲线含有7个浓度,用VCM与IS峰面积的比值对万古霉素浓度进行线性拟合,见表2,用标准曲线反算出的对照品血浆的VCM浓度见表3。
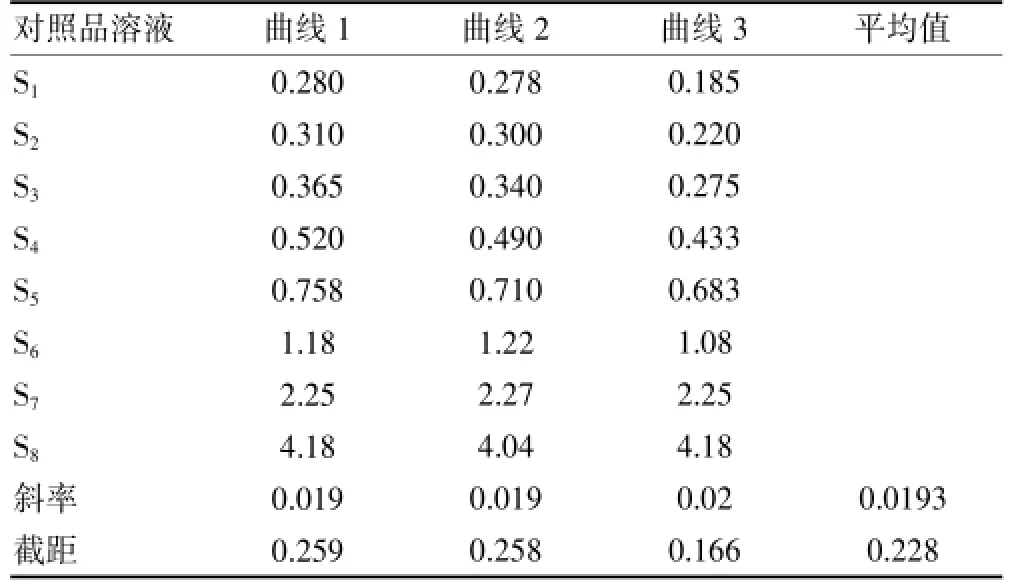
表2 万古霉素与IS峰面积比及回归曲线参数
标准曲线包括了7个浓度,n=7,每个对照品浓度测定的次数均为3次,m=3,N为测定标准血浆溶液的总次数,N=m×n=21;xi为第i个标准血浆溶液的浓度,为7个标准血浆浓度的理论平均值,x=49.5 μg/mL;am为标准曲线的斜率;bm为标准曲线的截距;i为对照溶液的序数(i=1,2,…,N),j为测定血浆标准溶液的序数(j=1,2,…,N)。
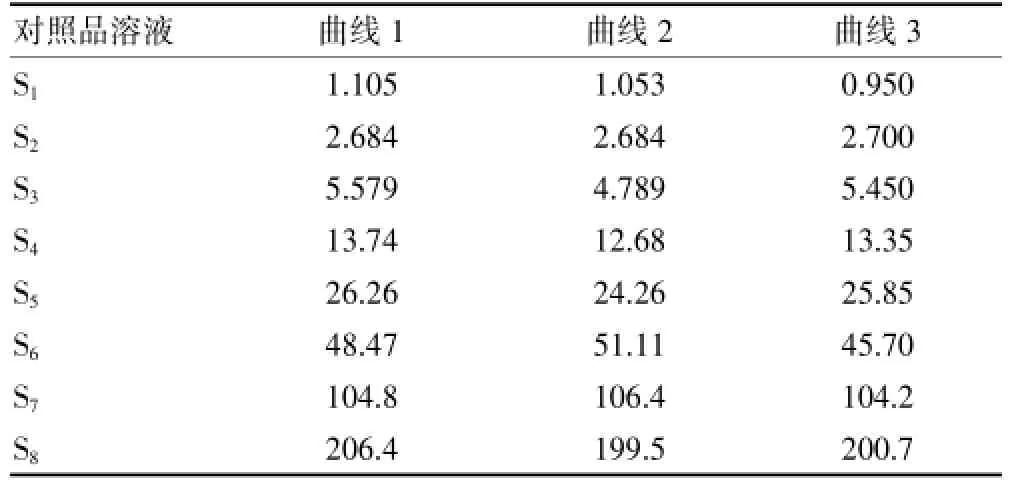
表3 用拟合曲线计算出每个对照品血浆万古霉素的浓度(μg/mL)
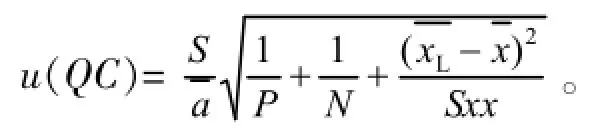
u(xL)=0.00486 μg/mL,u(xH)=0.006 71μg/mL则曲线拟合引入的相对标准测量不确定度为:
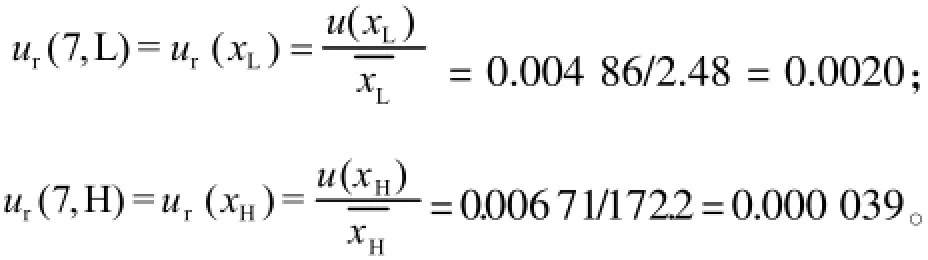
2.7.7温度、对照品纯度和样品不均匀性引入的不确定度
实验室温度变化控制在±2°C以内,并且标准曲线和待测样品在同一室温下制备和测定,温度引入的不确定度可以忽略不计。由于对照品纯度测定未提供不确定度,引入的不确定度则不予考虑。本实验中的样品为血浆,使用前经过充分混匀,由样品不均匀性引入的不确定度可以忽略。测量不确定度各分量大小的统计直方图见图2。
2.8标准测量不确定度的合成及扩展
2.8.1标准测量不确定度的合成
依据不确定度传播规律对各相对标准测量不确定度进行合成公式为:
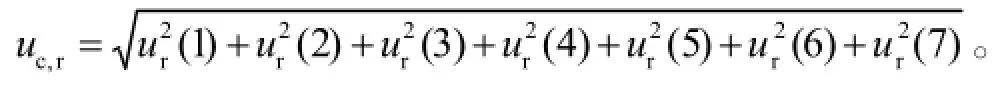
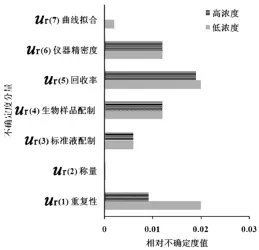
图2 不确定度分量的统计直方图
万古霉素质控样品的合成标准测量不确定度分别为:
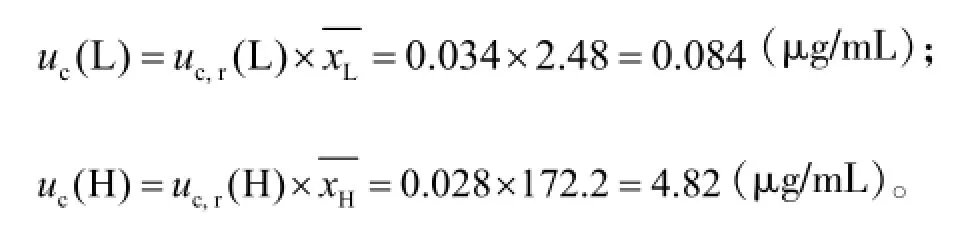
2.8.2标准测量不确定度的扩展
用简易评定法,对应的置信概率P=95%(k=2),则扩展不确定度分别为:
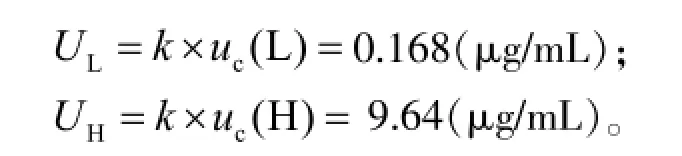
2.9测定结果的表示
当置信概率P=95%(k=2),血浆中万古霉素低、高浓度质控的测定结果可以分别表示为(2.480± 0.168)μg/mL和(172.20±9.64)μg/mL。
3 讨论
本试验选取天坛医:神经外科开颅术后使用万古霉素的患者。给药方式为VCM 1.0 g静脉内输注1 h,其后以125 mg/h(3 g/d)匀速持续静脉输注72 h。采集静脉血标本,用高效液相色谱法测定VCM浓度,样品的浓度在3.4~103 μg/mL之间。
本文分析了HPLC法测定血浆中VCM浓度的实验步骤和流程确定了不确定度的来源,根据规范、指南[7-9]并参考文献[10-11],将不确定度各分量进行量化,最后进行合成并扩展。高、低质控浓度回收率引入的不确定度差别很大。表明在进行萃取操作时,为了减小误差,应当制订萃取的标准操作规程并严格执行。生物样品配制引入的不确定度均较大,这与配制样品使用移液器的精密度及使用次数有关。
线性回归对低浓度的影响远大于高浓度,因此线性回归时选择合适的权重以保证低浓度测定的准确性十分必要。由于实验室温度可以控制,而且标准曲线和待测样品需要同时配制,温度变化小,温度引入的不确定度可以忽略不计。样品不均匀性引入的不确定度在固体样品中通常需要评定,而本实验中的样品均为液体,此项可忽略[5-7,9]。
综上所述,用高效液相紫外检测法测定血清中VCM含量的不确定度在高浓度时主要由生物样品配制、回收率和基质效应引入,在低浓度时主要由重复性、生物样品配制、基质效应和回收率引入。
[1]黄晓会,刘燕,张健.HPLC法测定人血清中万古霉素及去甲万古霉素浓度及临床应用[J].中国药物应用与监测,2014,11(2):92-94.
[2]Hu MW,Anne L,Fomi T,et a1.Measurement of vancomycin On rena11y impaired patient samp1es using a new high· performance 1iquid chromatographymethod with vitamin B12interna1 standard:comparison of high-performance 1iuid chromatography,emit,and f1uorescence po1arization immnnoassay methods[J].Therapeutic Drug Monit,1990,12(6):562-565.
[3]丰嘉驹,顾嘉钦.高效液相色谱法测定去甲万古霉素血药浓度[J].中国医:药学杂志,2000,20(8):458-459.
[4]涂厉标,王真,李旭梅.高效液相色谱法测定血清去甲万古霉素与万古霉素浓度[J].医药导报,2007,26(5):485-487.
[5]戴智勇,李新中,尹桃,等.反高效液相色谱法测定人血浆中万古霉素浓度[J].中国抗生素杂志,2002,27(10):599-600.
[6]臧慕文.分析测试不确定度的评定与表示(I)[J].分析实验室,2005,24(11):74-79.
[7]测量不确定度评定与表示[S].JJF1059.1-2012,2012.
[8]检测和校准实验室能力认可准则[S].CNAS-CL01,2006.
[9]中国合格评定国家认可委员会.CNAS-GL06:2006化学分析中不确定度的评估指南[S].北京:中国计量出版社,2006.
[10]王猛猛,贺敏,过林,等.LC-MS/MS法测定人血浆中吡啡尼酮的浓度及其方法不确定度评定[J].中国药房,2015,26(8):1056-1059.
[11]过林,裘福荣,王猛猛,等.LC-MS/MS法测定人血浆中双氯芬酸浓度的不确定度评定[J].安徽医药,2015,19(4):627-631.
Uncertainty evaluation on the determination of vancomycinin human Plasma by HPLC method
WANG JiaqingZHAO ZhigangMEI Shenghui
Department of Pharmacy,Beijing Tiantan Hospita1,Capita1 Medica1 University,Beijing100050,China
R927.1
A
1673-7210(2016)04(a)-0030-05
北京市重点实验室课题(2014DTYW03)。
王佳庆(1984-),女,硕士,主要从事临床医学和药物分析研究。
梅升辉(1983-),男,博士,主要从事药物化学和药物分析研究。
(2015-12-24本文编辑:赵鲁枫)



