陈超洪 王 优 李承燕 刘 玲 敖 当
广东医科大学附属医院儿童医学中心,广东湛江 524000
发育性癫痫性脑病(developmental and epileptic encephalopathy,DEE)是一组以发育障碍、难治性癫痫为特征的疾病[1]。其中DEE-95 与磷脂酰肌醇聚糖锚定生抑合成S 类(phosphatidylinositol glycan anchor biosynthesis class S,PIGS);OMIM*610271)基因突变相关,是一种常染色体隐性遗传病,表现为婴儿期起病的难治性癫痫[2],有多种癫痫发作类型[3],以严重发育迟缓、肌张力减退、面部异常(拱形眉、杏仁眼、鼻梁低平、长人中、宽舌)为特征[4],少数患儿共患孤独症谱系障碍[5]。本研究通过对表型相近的2 例难治性癫痫患儿进行基因检测,明确其致病原因,为临床诊断和遗传咨询提供参考。
1 对象与方法
1.1 研究对象
收集2021—2022 年因“反复抽搐”就诊广东医科大学附属医院门诊的2 例患儿的临床资料及家族史。
病例1:8 岁女性患儿,因“反复抽搐7 年余”于广东医科大学附属医院(以下简称“我院”)门诊就诊。生后5 个月出现高热抽搐,表现为双眼上翻,面色发绀,双手握拳,四肢强直抖动,持续约2 min 后自行缓解。随后出现无热抽搐,表现同前,2~3 次/周,外院予丙戊酸(25 mg/kg)联合托吡酯(5.4 mg/kg)抗癫痫治疗后无发作2 年,遂自行停药。停药后半年再次出现抽搐,发作形式同前,持续1~2 min 后自行缓解,外院予单用丙戊酸治疗,仍有抽搐,1~3 次/月,至我院门诊进一步确诊后,予丙戊酸(25 mg/kg)联合维生素B6(10 mg/kg)治疗,目前1 年无发作。生长发育落后,4个月抬头,12 个月能坐,1岁11 个月扶走,3 岁半能独立行走。目前可独立行走,摇摆步态,语言落后,仅发“啊啊、吧吧”叠音,能执行“再见”等简单指令,不能自行进食。评估发育商26,其中大运动35 分,精细运动25 分,认知26 分,语言15 分,社交25 分。
病例2:6 岁女性患儿,因“反复抽搐5 年余”于我院门诊就诊。患儿生后6 个月无明显诱因出现抽搐,表现为双眼凝视,面色发绀,双手握拳,四肢强直,持续约3 min 后自行缓解。每月发作4~15 次不等,发作间期神志清楚。外院予丙戊酸(26 mg/kg)及托吡酯(6.0 mg/kg)抗癫痫治疗,仍每月发作2~8 次不等。至我院门诊进一步确诊后,予丙戊酸(25 mg/kg)、托吡酯联合维生素B6(15 mg/kg)治疗,抽搐发作较前好转,1~2 次/年。生长发育落后,5 个月抬头,11 个月能爬,1 岁10 个月独坐。目前不能独立行走,语言落后,仅发“啊啊”叠音,不能执行简单指令,不能自行进食。评估发育商14 分,其中大运动15 分,精细运动15 分,认知16 分,语言0 分,社交15 分。
2 例患儿查体反应迟钝,眼神呆滞,特殊面容(拱形眉、杏仁眼、眼距宽、鼻梁低平、长人中、宽舌),心、肺、腹未见异常,四肢肌张力减低,四肢肌力正常,双侧膝腱反射正常引出,病理征阴性,均未发现疝气或脊柱、四肢骨骼明显发育异常。2 例患儿既往史、出生史无特殊。父母表型均无异常,母亲孕期无异常,否认近亲结婚,否认家族中其他成员有癫痫病史。本研究已获得患儿家属知情同意及我院伦理委员会审查批准(YJYS2022173)。
1.2 研究方法
1.2.1 实验室检查 对2 例患儿进行血常规、尿常规、肝肾功能、心肌酶谱、血生化、血氨、同型半胱氨酸、血清铜蓝蛋白、血乳酸等,听力筛查、血尿串联质谱及脏器彩超、视频脑电图监测、头颅磁共振检测。
1.2.2 全外显子组测序 抽取2 例患儿及其家族成员静脉血各2 ml,外送深圳安吉康尔公司进行全外显与测序,提取全血基因组DNA,经PCR 扩增、测序,过程采用FastQC 软件做质量评估,使用BWA 软件进行序列比对。
1.2.3 Sanger 测序验证 由安吉康尔公司进行引物设计,基于转录本NM_033198.4,采用梯度PCR,PCR 产物经纯化后构建测序反应,使用ABI 3730XL 全自动测序仪进行测序验证。
1.2.4 突变分析 采用美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南进行基因突变筛选及致病评级。
2 结果
2.1 实验室检查
2 例患儿的血常规、生化检查、血尿串联质谱筛查及染色体核型分析、脏器彩超、听觉诱发电位等未见异常,头颅磁共振示存在不同程度的异常(图1)。病例1 视频脑电图提示双侧额极稍多量慢波不同步发放;睡眠期右侧额、中央、顶区或额中央区、中线中央区少量尖波散发(图2A)。病例2 视频脑电图提示双侧额极不对称,右侧额极多量中-极高波幅混合慢波发放;睡眠期左侧额、额中央区、前颞、额极少量棘波、棘慢波散发;睡眠期前头部为主或广泛性慢波发放(图2B)。
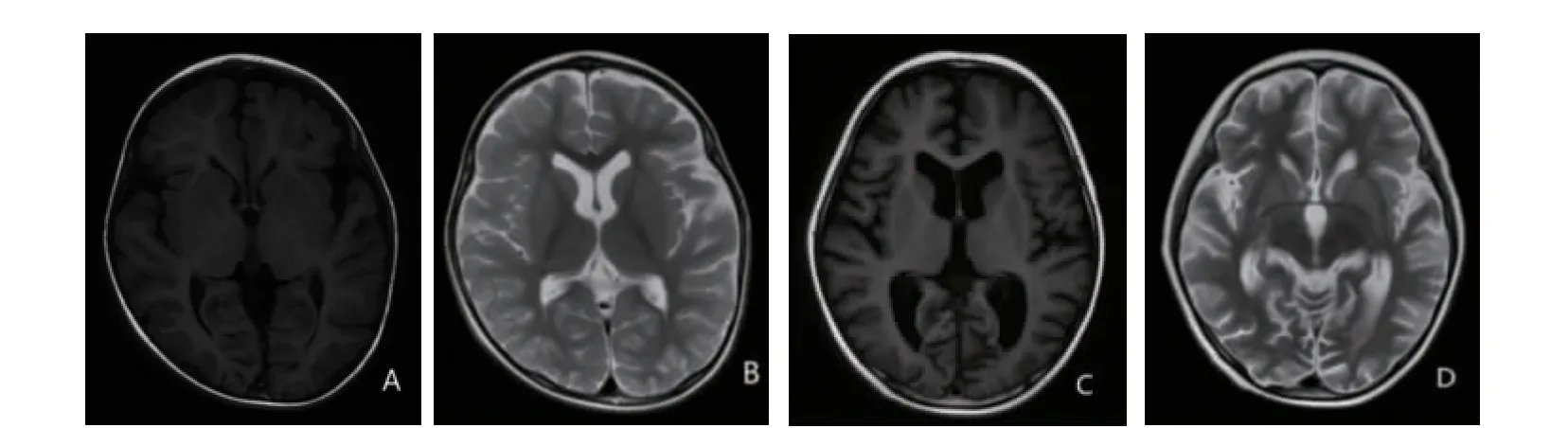
图1 PIGS 基因突变致DEE-95 患儿头颅磁共振成像横断位图像
2.2 基因检测结果
全外显子基因和拷贝数突变检测示2 例患儿基因拷贝数变异检测结果阴性,PIGS 基因c.1141_1164dup(p.Asp381_ Val388dup)纯合突变(图3),此突变导致第381 号氨基酸天冬氨酸到第388 号氨基酸缬氨酸发·重复(p.Asp-381_Val388dup),该突变在参考人群基因频率数据库(gnomAD)中最小等位基因频率未见记录(PM2),为非重复区框内插入/缺失或终止密码子丧失导致的蛋白质长度变化(PM4),根据ACMG 指南(PM2+PM4),该突变的致病性为意义不明确。Sanger 测序检测到父母均携带c.1141_1164dup(p.Asp381_ Val388dup)杂合突变,PIGS 基因突变所致DEE95 符合常染色体隐性遗传(图4)。
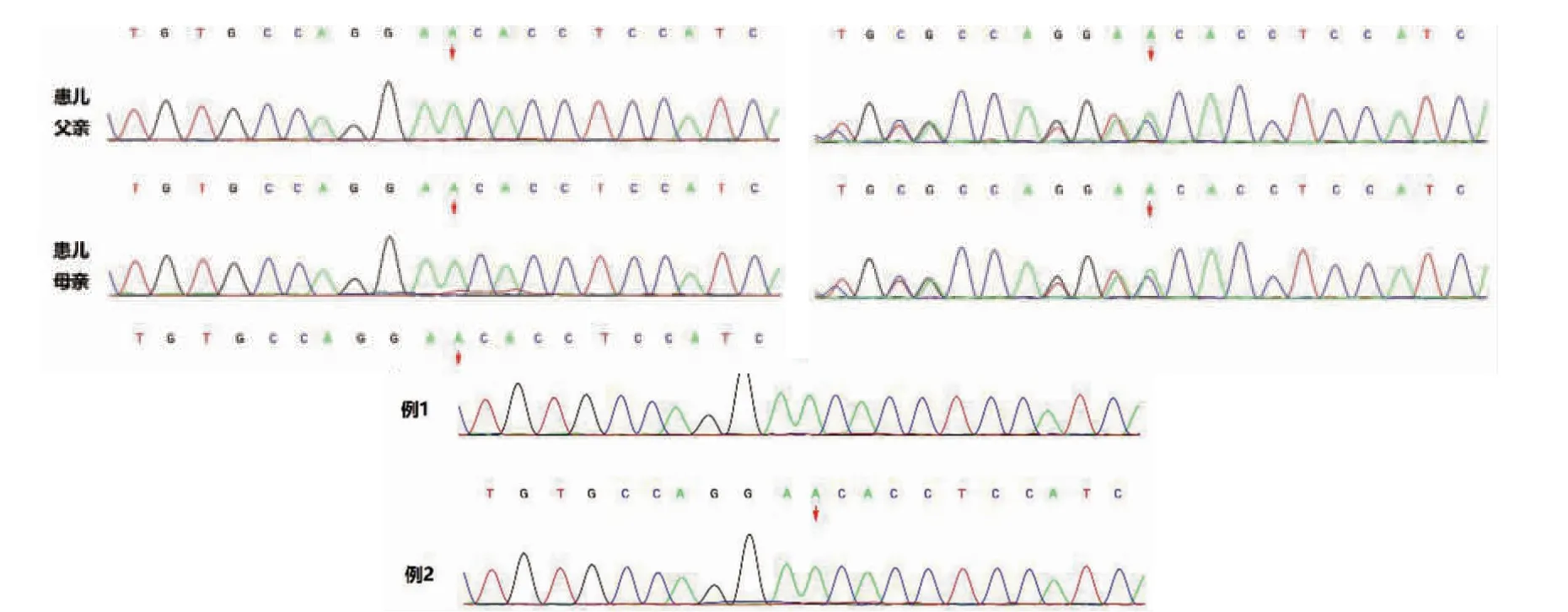
图3 PIGS 基因突变致DEE-95 患儿及父母Sanger 测序
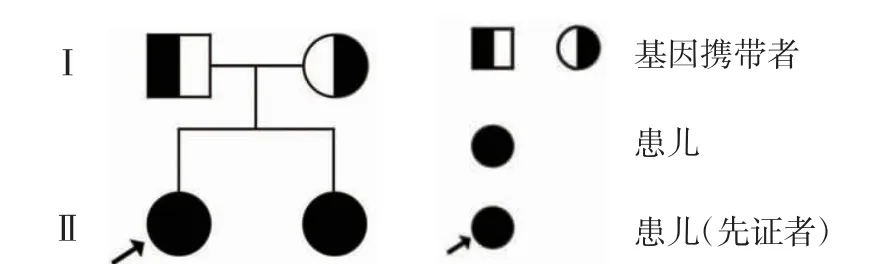
图4 PIGS 基因突变致DEE-95 家系图谱
3 讨论
PIGS 基因参与糖基磷脂酰肌醇(glycosylphosphatidylinisotol,GPI)锚定生物合成过程[6],与GAA1、GPI8、PIGT 和PIGU 组成GPI 转酰胺酶[7],在突触功能、神经发育和可塑性中发挥作用[8]。PIGS 蛋白在小脑中表达最高[9],是识别GPI 附着信号所必需的[10]。PIGS 基因突变导致GPI 生物合成缺陷,从而出现遗传性糖基磷脂酰肌醇锚定蛋白缺陷(inherited glycosylphosphatidylinisotol-anchored protein deficiencies,IGD)[11]。IGD 是先天性糖基化障碍一类,占所有神经发育障碍病因的0.15%[12-13],包括不同程度的智力/发育障碍、癫痫、肌张力减退、共济失调、先天性结构畸形等[14-16]。研究显示,GPI 转酰胺酶亚基等位基因的功能丧失会导致DEE[16-17]。
本研究家系中2 例患儿均在婴儿期出现频繁癫痫发作,伴严重发育落后和特殊体征(拱形眉、杏仁眼、眼距宽、鼻梁低平、长人中、宽舌),异常视频脑电图及头颅磁共振,单纯抗癫痫药物治疗效果欠佳,联用维生素B6治疗可改善癫痫发作。经全外显子组测序发现PIGS 基因纯合突变,该突变使所编码的蛋白失去原有功能,导致细胞膜表面的锚定蛋白合成缺陷,理论上有致病性,且该突变人群携带率低,被Clinvar数据库报道为致病(Pathogenic,https://www.ncbi.nlm.nih.gov/clinvar/variation/1119956),综合2 例患儿临床表型特点,符合PIGS 突变致DEE-95,具有临床致病意义,需要更进一步的实验研究。检索国内外相关文献,本研究2 例患儿与文献报道中15 例DEE-95 患儿均存在重叠的临床表型,包括严重发育迟缓、婴儿期起病的难治性癫痫、肌张力减退、面容异常等,脑电图特征多表现为多灶性棘波和高波幅慢波,颅脑磁共振异常以额颞叶及小脑发育不全多见。15 例患儿中部分可伴随脊柱四肢骨骼畸形、视听功能障碍、心脏和泌尿系统发育畸形[18-20]。本研究中2 例患儿多系统受累情况较国外文献报道相对少,是否存在人种差异待进一步探索。目前DEE-95 治疗主要是多种抗癫痫药物联合使用[21-22],国外曾报道生酮饮食和维生素B6对癫痫控制有效[23-26],本研究1 例患儿加用维生素B6治疗达到无发作。
综上所述,PIGS 基因突变所致DEE-95 罕见,该病可累及神经、骨骼、视觉、听觉、泌尿、心血管等多器官系统,对于婴儿期起病的难治性癫痫,同时合并严重发育迟缓、肌张力减退、面容异常、颅脑磁共振异常等多种表型的患儿,建议行遗传学检测,对早期识别、精准诊断和遗传咨询有重要意义。现有治疗方案中可以观察到维生素B6治疗对于控制癫痫发作有积极作用。



