宋雯妍,张瀚文,吴澳迪,张丽燕,刘 照,叶桐桐,陈创夫,盛金良
(石河子大学动物科学院,石河子 832000)
猪繁殖与呼吸综合征又称猪蓝耳病,是由猪繁殖与呼吸综合征病毒(porcine reproductive and respiratory syndrome virus, PRRSV)感染而导致妊娠母猪在怀孕晚期出现流产,育肥猪食欲不振,体重减轻,以及各日龄猪呼吸系统紊乱的疾病[1-4]。PRRSV是单股正链RNA病毒,属于动脉炎病毒科动脉炎病毒属,其结构为二十面体,具立体对称性,病毒衣壳内含有不分节段的基因组 RNA,全长大小约为 15 kb,编码至少8 种结构蛋白与14 种非结构蛋白[5-6]。GP5 蛋白又称E蛋白,是由病毒 ORF5 开放阅读框编码的主要糖基化结构的跨膜蛋白,其特点是具有较好的免疫原性,并能够诱导机体产生中和抗体,且该蛋白在体内及体外试验中,均能诱导细胞发生凋亡[7-8]。此外GP5蛋白与 M 蛋白可形成 GP5/M 异源二聚体,于病毒的装配起到重要作用,并介导PRRSV对靶细胞的吸附过程[9-10]。鉴于GP5蛋白于病毒的生命周期中发挥着重要生物学功能,因此该蛋白是研发抗猪蓝耳病药物的理想靶标蛋白。
纳米抗体(Nb)是研究人员基于骆驼等驼科动物以及鲨鱼等软骨鱼体内的重链抗体可变区片段上所研发的一种单域抗体,其仅由重链可变区构成,长4 nm,直径2.5 nm,呈椭圆形,其大小仅是传统抗体的1/10,是目前所知最小且具备活性的抗原结合蛋白[11-12]。纳米抗体因具备体积小、亲和力高、稳定性与组织穿透性强等优势,近年来其于病毒感染治疗领域的相关研究不断深入。目前已有针对SARS-冠状病毒-2、艾滋病病毒、甲型流感病毒等多种病毒的纳米抗体报道,研究结果显示,这些纳米抗体对病毒复制的抑制效应较好,可作为治疗病毒感染的理想工具。
本研究基于研发PRRSV GP5蛋白纳米抗体,并探究其在细胞内对病毒复制的抑制效力,目前针对此蛋白纳米抗体的相关研究尚未见报道。因此,将GP5蛋白经原核表达并纯化后免疫羊驼,利用噬菌体展示技术最终筛选出PRRSV-GP5蛋白的高特异性纳米抗体,并将其转染入细胞内以验证该纳米抗体对PRRSV复制与转录产生的影响,为抗PRRSV新型药物的研发奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌种、表达载体、试验动物 大肠杆菌 DH5α、BL21(DE3)、TG1及pET-32a载体、pCANTAB-5E、pcDNA3.1(-)载体均由石河子大学兽医病理学实验室保存;健康成年母羊驼1只,饲养于石河子大学动物科技学院。
1.1.2 细胞与毒株 Marc-145细胞与PRRSV毒株均由石河子大学动物科技学院人兽共患病实验室保存。
1.1.3 主要试剂 反转录试剂盒、PCR反应试剂、 DNA Marker、 Protein Marker、 Ni-NTA Resin、 Anti-His Mouse Monoclonal Antibody、 Goat Anti-Mouse IgG (H+L), HRP Conjugate均购自北京全式金生物技术有限公司;T-Vector pMD19(Simple)、 DNA Ligation Kit Ver.2.1、 限制性内切酶BamHⅠ、HindⅢ、PstⅠ、NotⅠ均购自于宝日医生物技术(北京)有限公司;Anti-M13 Antibody (HRP)购自北京义翘神州科技股份有限公司;E-tag Antibody(HRP)购自金斯瑞生物科技股份有限公司;无内毒素质粒大提试剂盒购自天根生化科技(北京)有限公司;Lipofectamine3000购自美国英杰生命技术有限公司;Goat Anti-Llama IgG H&L(HRP)为进口试剂。
1.2 方法
1.2.1 PRRSVORF5基因重组质粒的构建 根据GenBank中PRRSV XJzx1-2015分离株的GP5序列(AQV08435.1)进行生物信息学分析,去除该基因中的信号肽及跨膜区序列,并针对优化后的截短序列设计引物(表1),以实验室保存的PRRSV阳性病料为模板,使用PCR技术扩增ORF5基因,并通过限制性内切酶BamHⅠ和HindⅢ将ORF5基因克隆至原核表达载体pET-32a中,得到重组质粒pET-32a-GP5,后转化至DH5α感受态细胞。
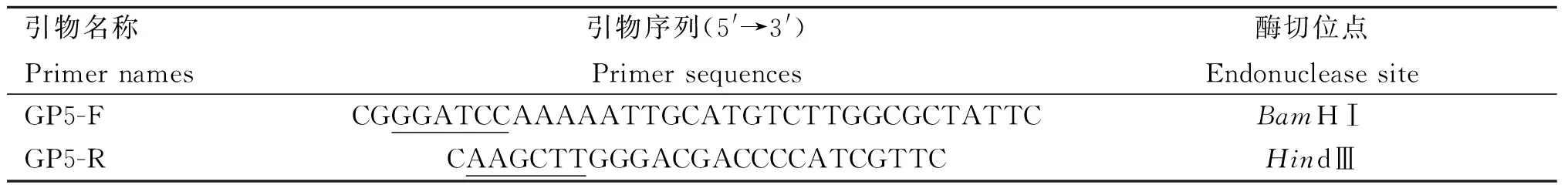
表1 构建重组质粒pET-32a-GP5的引物序列Table 1 Primer sequence for constructing recombinant plasmid pET-32a-GP5
1.2.2 PRRSV GP5蛋白的诱导表达及纯化 将经双酶切及测序正确的重组质粒,转化入大肠杆菌DE3(BL21)感受态细胞中,挑单克隆入含有氨苄抗性的LB培养基中扩大培养,37 ℃,200 r·min-1培养至菌液OD600 nm值达到0.6时停止,取1 mL菌液作为诱导前对照,后于剩余菌液中加入1 mmol·L-1的IPTG进行诱导,37 ℃,200 r·min-1培养6 h,同时设置空载体对照,SDS-PAGE鉴定重组蛋白的表达。取诱导后菌液进行超声破碎(功率50 w,超声5 s,间隔6 s,90 次·周期-1),收集上清与沉淀进行可溶性分析并使用镍柱亲和层析法对重组蛋白进行纯化,后Western blot鉴定。
1.2.3 动物免疫 首次免疫前,对羊驼进行采血以作阴性对照,后将纯化好的1 mL GP5重组蛋白与等量弗氏完全佐剂混合乳化,于羊驼背部体表进行多点皮下注射。此后每隔14 d使用0.5 mL重组蛋白与等量弗氏不完全佐剂乳化对羊驼进行加强免疫,直至三免结束,分别于免疫第0、14、28、42 天时采血,使用间接ELISA方法检测羊驼体内抗体效价,随后采集羊驼全血,分离淋巴细胞以备后续试验。
1.2.4 VHH基因的扩增 提取淋巴细胞RNA,反转录后参考文献[13]中的引物序列(表2)进行巢式PCR扩增出VHH片段。首先使用引物CALL001、CALL002进行第一轮PCR扩增,反应条件:98 ℃预变性30 s;98 ℃变性10 s,55 ℃退火30 s,72 ℃延伸25 s,共28 个循环;最后72 ℃延伸 5 min,PCR产物经1.5%琼脂糖凝胶电泳鉴定,并对位于700 bp的条带进行胶回收以用于第二轮PCR的模板,使用引物VHH-F及VHH-R进行VHH片段的扩增。反应条件:98 ℃预变性 30 s;98 ℃变性10 s,55 ℃退火30 s,72 ℃延伸15 s,共18个循环;最后72 ℃延伸 5 min,PCR产物经1.5%琼脂糖凝胶电泳鉴定,对位于400 bp的目标条带进行胶回收并于-20 ℃保存。
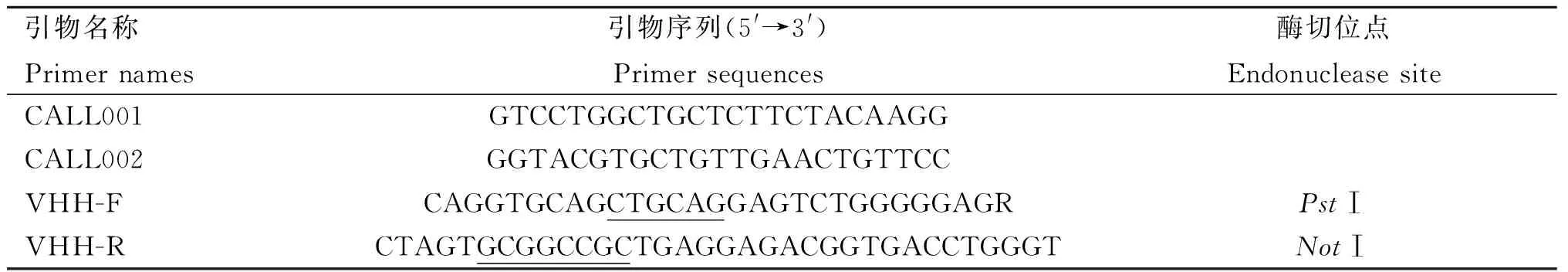
表2 扩增VHH片段所需引物Table 2 Primers required for VHH fragment amplification
1.2.5 VHH噬菌体展示文库的构建及鉴定 将VHH片段与pCANTAB 5E载体分别用限制性内切酶PstⅠ和NotⅠ于37 ℃双酶切1 h后使用T4连接酶于16 ℃连接16 h。取10 μL连接产物转化入100 μL大肠杆菌TG1感受态细胞中,次日用细胞刮刀收集培养皿中的所有菌落并充分混匀,即可得到VHH噬菌体展示文库。使用2×YT液体培养基进行10倍梯度稀释,加入等体积处于对数生长期的TG1溶液,混匀,37 ℃静置感染30 min,涂布于LB/AMP固体培养基中进行过夜培养,统计转化子的数量以计算库容。随机挑取培养皿中的阳性克隆进行菌液PCR鉴定并计算文库插入率。
1.2.6 VHH噬菌体展示文库的救援 取1 mL上一步制备的VHH噬菌体展示文库溶于100 mL 2×YT/AMP液体培养基中,37 ℃,200 r·min-1培养至菌液OD600 nm值达到0.6时停止,加入200 μL M13K07辅助噬菌体(6.14×1012PFU·mL-1)进行救援,混匀,37 ℃静置30 min后于37 ℃,200 r·min-1,振荡培养40 min。离心后弃上清,菌体沉淀用500 mL 2×YT/AMP 液体培养基重悬,37 ℃,200 r·min-1,振荡培养14 h。14 000 r·min-1,离心后收集上清,加入1/5体积提前预冷的PEG/NaCl溶液,混匀后于冰上静置6 h。离心后收集沉淀,用1 mL的无菌PBS(1×)重悬沉淀后于4 ℃摇床过夜孵育,完全溶解噬菌体颗粒。
1.2.7 PRRSV-GP5特异性纳米抗体的淘选 取纯化后GP5蛋白用碳酸盐包被液稀释至10 μg·mL-1,每孔100 μL加入至96孔酶标板中,同时设置PBS(1×)作为阴性对照,4 ℃包被过夜。弃去包被液,5%脱脂乳于37 ℃封闭2 h,PBST洗板。加入稀释至6.1×1010的重组噬菌体(“1.2.6”方法),100 μL·孔-1,37 ℃孵育2 h,洗板后加入新鲜配制的0.1 mol·L-1三乙胺溶液,100 μL·孔-1,37 ℃静置10 min后吸出洗脱液,并迅速加入等体积的1 mol·L-1Tris-HCl(pH=7.4)进行中和,对洗脱液中的重组噬菌体滴度进行测定,并计算回收率。取4 mL 培养至对数生长期的TG1溶液,加入400 μL洗脱的重组噬菌体溶液,混匀后于37 ℃静置30 min,加入16 mL的2×YT/AMP 液体培养基,37 ℃,200 r·min-1培养至菌液OD600 nm值达到0.6时停止,加入100 μL M13K07辅助噬菌体救援,按照“1.2.6”的方法对噬菌体粒子进行浓缩纯化,重复上述操作三次,完成对重组噬菌体的三轮淘选。
1.2.8 重组纳米抗体的诱导表达与粗提物获取 取重组噬菌体第三轮淘选的洗脱液,测定滴度后从LB-AMP固体培养基中随机挑取96个单克隆,过夜培养。次日各菌液取10 μL培养物转接至1 mL TB培养基,37 ℃,200 r·min-1培养至菌液OD600 nm值达到0.6时加入终浓度为1 mmol·L-1的IPTG诱导12 h。离心后吸弃上清液,菌体反复冻融三次后使用无菌PBS(1×)重悬,离心后收集上清即获得可溶性纳米抗体粗提物。
1.2.9 重组纳米抗体的间接ELISA检测 将纯化后的GP5蛋白稀释至10 μg·mL-1,100 μL·孔-1加入至96孔酶标板中,共包被同时设置PBS(1×)作为阴性对照,4 ℃包被过夜。次日弃包被液,用5%脱脂乳于室温封闭2 h,PBST洗板。将可溶性纳米抗体粗提物用5%脱脂乳1∶1稀释后,每孔加入100 μL,37 ℃孵育1 h。PBST洗板,分别加入Anti-M13抗体和Anti-E-tag抗体以作二抗,100 μL·孔-1,37 ℃孵育1 h后进行显色,加入ELISA终止液终止反应后于酶标仪测取OD450 nm并分析数据。
1.2.10 PRRSV-GP5 特异性纳米抗体序列分析 将与Anti-M13抗体和Anti-E-tag抗体反应均为阳性的单克隆扩大培养,进行菌液PCR鉴定后阳性者送去测序,用DNAMAN软件比对测序结果,并根据纳米抗体的高变区序列归类。
1.2.11 PRRSV-GP5 特异性纳米抗体对PRRSV增殖的影响 以所筛PRRSV-GP5特异性纳米抗体所对应的菌液为模板,使用表3所示的引物进行PCR扩增,并通过限制性内切酶SacⅠ和Hind Ⅲ将扩增出的纳米抗体基因克隆至真核表达载体pcDNA3.1(-)中,得到重组质粒pcDNA3.1(-)-Nb1与pcDNA3.1(-)-Nb2,后转化至DH5α感受态细胞。次日,随机挑取转化成功的单克隆进行菌液PCR验证及双酶切鉴定,二者均正确者送去测序。
将含有构建成功质粒的菌液进行扩大培养并使用无内毒素质粒大提试剂盒进行质粒提取,后使用Lipofectamine3000将重组质粒转染至Marc-145细胞中,经Western blot及荧光定量PCR(引物见表4)检测Nb mRNA的相对表达量来鉴定重组质粒于细胞中的表达情况。
将pcDNA3.1(-)-Nb1、pcDNA3.1(-)-Nb2分别转染至Marc-145细胞中,取1 MOI PRRSV 接种各组细胞,同时设置空细胞接毒组作为阴性对照,37 ℃,5% CO2培养箱中共孵育1 h(期间每30 min轻柔晃动细胞培养板),弃去毒液,1×PBS洗两遍,于各组0 h细胞样品中加入TRIZOL裂解后置于-80 ℃保存备用,剩余各组加入含3% FBS的DMEM培养基,继续置于37 ℃,5% CO2培养箱中进行培养,后分别于24、36、48、60、72 h收集各组细胞样本,进行RNA提取后反转录,并使用表4所示的引物检测PRRSVORF7基因mRNA相对表达量的变化以判定各组纳米抗体在不同时间段对病毒复制所产生的影响。
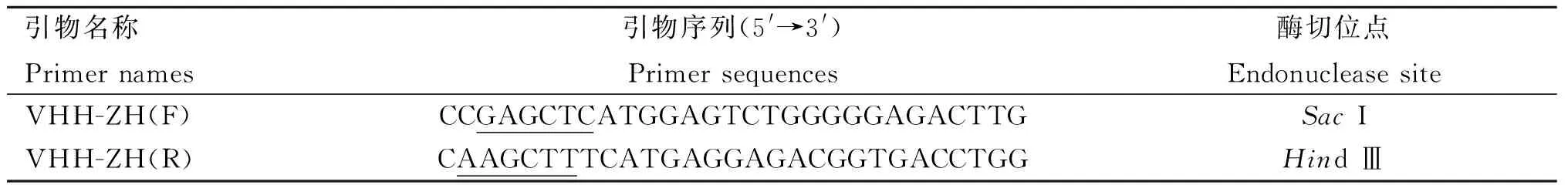
表3 构建重组质粒pcDNA3.1(-)-Nbs的引物序列Table 3 Primer sequence for constructing recombinant plasmid pcDNA3.1(-)-Nbs
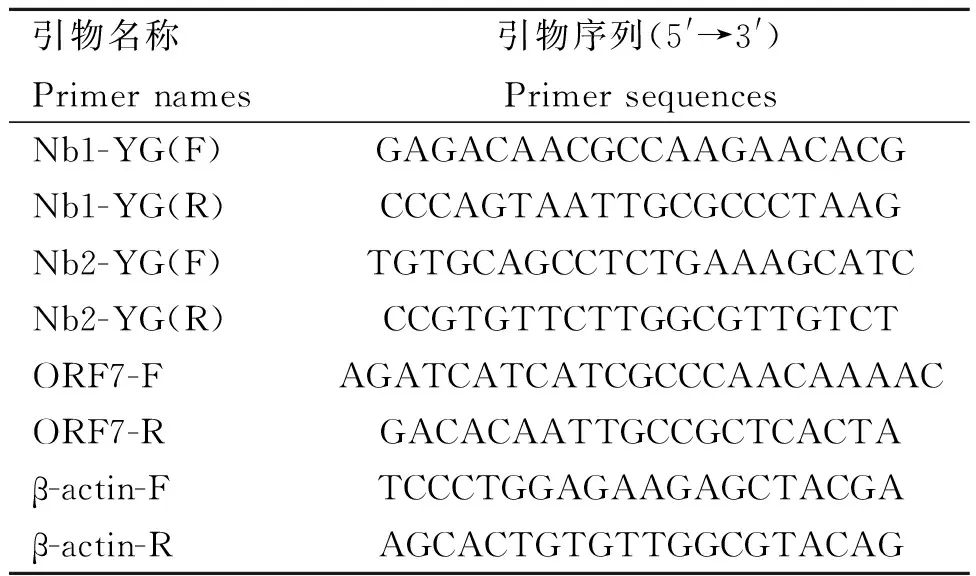
表4 荧光定量PCR引物序列Table 4 Fluorescence quantitative PCR primer sequences
2 结 果
2.1 pET-32a-GP5重组质粒的构建
取实验室保存的PRRSV阳性病料,该病料据文献 [14]记载,内含毒株为野毒株,且通过对其进行遗传变异分析可知,该毒株与香港PRRSV毒株的遗传进化关系较为接近,推测其与经典毒株相比,毒性有所增强,另外,将该毒株的ORF5基因序列与19株PRRSV美洲型毒株进行序列比对,发现该基因针对不同毒株的广谱性较好。因此,本试验使用PCR技术对PRRSVORF5基因进行扩增,得到一条片段大小为216 bp的目的条带(图1A),与预期目的基因片段大小一致。回收目的条带后与原核表达载体pET-32a分别通过限制性内切酶BamH Ⅰ和Hind Ⅲ双酶切后进行连接,构建出重组质粒pET-32a-GP5,随后进行菌液PCR验证(图1B)与双酶切鉴定(图1C),并送去测序。结果表明,成功构建重组质粒pET-32a-GP5。
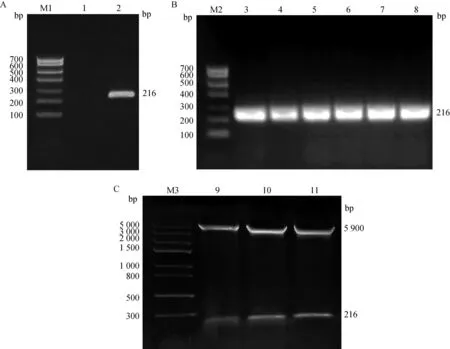
A. PRRSV ORF5基因的PCR扩增;B. 重组质粒pET-32a-GP5的PCR鉴定;C. 重组质粒pET-32a-GP5的双酶切鉴定;M1. DL700 DNA相对分子质量标准;1. 阴性对照;2. ORF5基因PCR扩增产物;M2. DL700 DNA相对分子质量标准;3~8. 菌液PCR扩增产物;M3. DL5000 DNA相对分子质量标准;9~11. pET-32a-GP5重组质粒 A. PCR amplificaiton of PRRSV ORF5 gene; B. Identification of pET-32a-GP5 by PCR; C. Identification of pET-32a-GP5 by double enzyme digestion; M1. DNA marker DL700; 1. Negative control; 2. PCR amplification product of ORF5 gene; M2. DNA marker DL700; 3-8. PCR amplification products of bacterial fluid; M3. DNA marker DL5000; 9-11. Recombinant pET-32a-GP5图1 pET-32a-GP5重组质粒的构建Fig.1 Construction of pET-32a-GP5 recombinant plasmid
2.2 PRRSV GP5蛋白的诱导表达及纯化
对含有pET-32a-GP5的重组质粒的菌液于1 mmol·L-1的IPTG诱导表达6 h后,将空载体对照、诱导前对照以及诱导后的重组蛋白均进行SDS聚丙烯酰胺凝胶电泳,结果显示空载体及诱导前对照无重组蛋白表达,诱导后重组蛋白于28 ku处有明显的蛋白条带产生(图2A),与预期大小一致。经可溶性分析,重组蛋白于包涵体沉淀中表达(图2B),使用镍柱亲和层析法过柱纯化GP5重组蛋白,最终收集到纯度较好的2.16 mg·mL-1的纯化后蛋白(图2C)。
2.3 羊驼免疫
采用间接ELISA法对GP5蛋白免疫羊驼血清抗体进行检测,结果显示三免14 d后,羊驼体内IgG抗体效价高达1∶819 200,对比阴性血清对照组,抗体水平显着增加(图3)。
2.4 VHH基因片段的扩增
提取羊驼全血淋巴细胞RNA,反转录后首先使用引物CALL001、CALL002进行第一轮PCR扩增,分别得到一条片段大小为900 bp的条带与另一条大小为700 bp的目的条带(图4A),胶回收位于700 bp的条带以用于第二轮PCR的模板,使用引物VHH-F及VHH-R对VHH片段进行扩增,最终位于400 bp处出现目的条带(图4B)。
2.5 VHH噬菌体展示文库的构建及鉴定
将VHH片段克隆至pCANTAB 5E载体并转化至大肠杆菌TG1感受态细胞中,得到库容量为2.93×107CFU·mL-1的VHH噬菌体展示文库,插入率为98%。
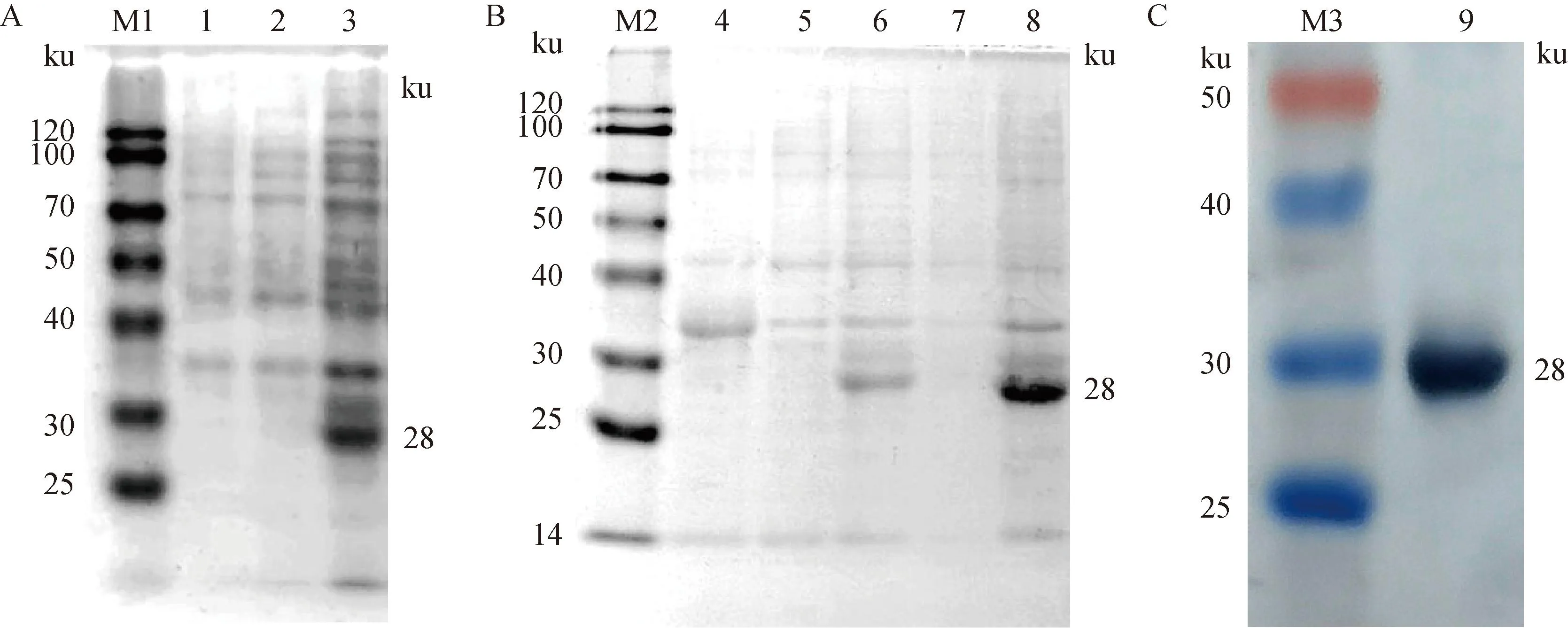
A.重组蛋白的SDS-PAGE鉴定;B. 重组蛋白的可溶性分析;C. 重组蛋白的Western blot鉴定;M1. 蛋白质相对分子质量标准;1. pET-32a空载体; 2. pET-32a-GP5诱导前;3. pET-32a-GP5诱导后;M2. 蛋白质相对分子质量标准;4. pET-32a空载体; 5. pET-32a-GP5诱导前; 6. pET-32a-GP5诱导后;7. 超声后上清;8. 超声后沉淀;M3. 蛋白质相对分子质量标准;9. pET-32a-GP5纯化 A. SDS-PAGE identification of recombinant protein; B. Solubility analysis of recombinant protein; C. Western blot identification of recombinant protein; M1. Protein molecular weight standard;1. pET-32a empty plasmid control; 2. pET-32a-GP5 before induction; 3. pET-32a-GP5 after induction; M2. Protein molecular weight standard; 4. pET-32a empty plasmid control; 5. pET-32a-GP5 before induction; 6. pET-32a-GP5 after induction; 7. Supernatant after ultrasound; 8. Precipitation after ultrasound; M3. Protein molecular weight standard;9. Purification of pET-32a-GP5图2 PRRSV GP5蛋白的诱导表达及纯化Fig.2 Inducible expression and purification of PRRSV GP5 protein
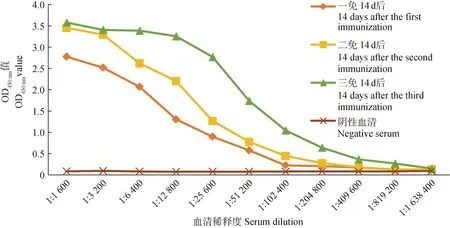
图3 羊驼血清中PRRSV GP5特异性抗体效价检测Fig.3 Detection of PRRSV GP5 specific antibody titer in alpaca serum
2.6 PRRSV-GP5特异性纳米抗体的淘选
将PRRSV-GP5蛋白作为包被抗原以筛出高亲和力与特异性纳米抗体。进行“吸附-洗脱-富集”共三轮淘选,通过测定每轮筛选过程中重组噬菌体的投入量与洗脱液里重组噬菌体的洗脱量,来计算回收率以评估PRRSV-GP5蛋白筛选过程中特异性噬菌体的富集情况。结果如表5所示,经三轮淘选后,重组噬菌体得到了有效富集,洗脱量与回收率均逐渐升高,其中,特异性噬菌体的洗脱量达2.48×108CFU·mL-1。
2.7 重组纳米抗体的间接ELISA检测
于重组噬菌体第三轮淘选滴度测定的固体培养基中随机挑取96个单克隆,经扩大培养与诱导表达后,制得可溶性纳米抗体粗提物,用于间接ELISA检测纳米抗体与PRRSV-GP5蛋白的反应原性。其中分别选取Anti-M13抗体和Anti-E-tag抗体作为不同的二抗进行试验检测,将OD值大于阴性对照三倍以上即判定为阳性,结果如图5所示。
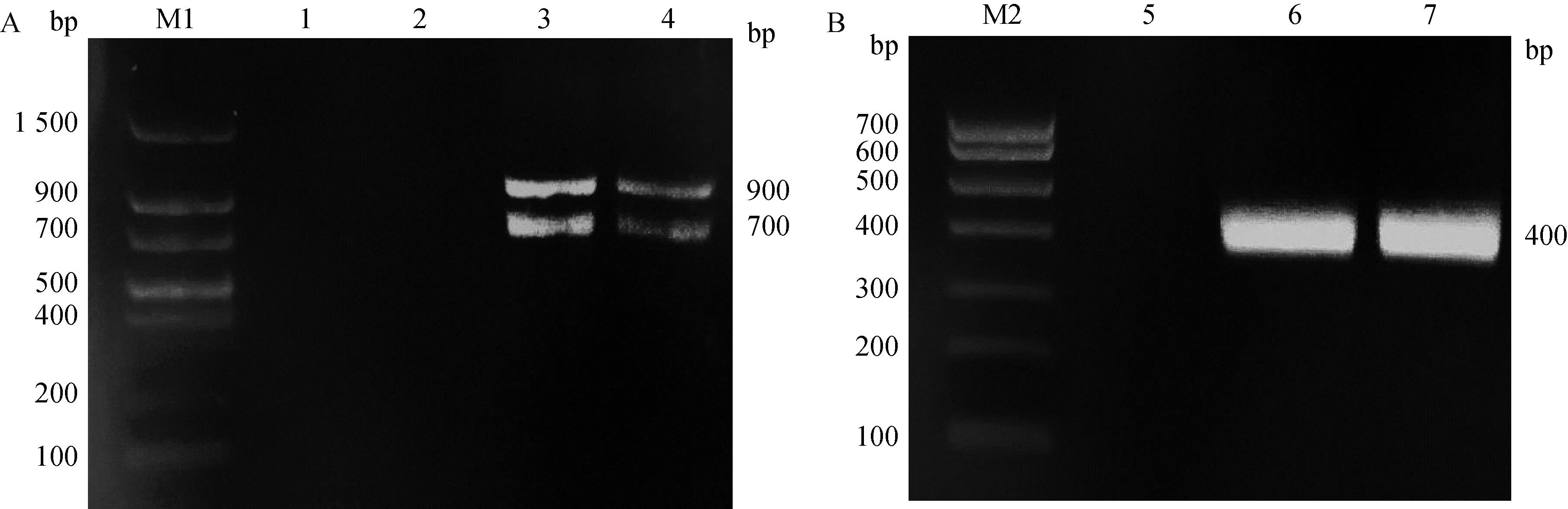
A. 巢式PCR第一轮扩增;B. 巢式PCR第二轮扩增;M1. DL1500 DNA相对分子质量标准;1、2. 阴性对照;3、4. 巢式PCR第一轮扩增产物;M2. DL700 DNA相对分子质量标准;5. 阴性对照;6、7,巢式PCR第二轮扩增产物 A. The first amplification round of nested PCR; B, The second amplification round of nested PCR; M1. DNA marker DL1500; 1, 2. Negative control; 3-4. The first round amplification products of nested PCR; M2. DNA marker DL700; 5. Negative control; 6-7. The second round amplification products of nested PCR图4 VHH基因片段的扩增Fig.4 Amplification of VHH gene fragment

表5 GP5蛋白筛选过程中特异性噬菌体的富集情况Table 5 Enrichment of specific phages during GP5 protein screening
2.8 PRRSV-GP5 特异性纳米抗体序列分析
将与Anti-M13抗体和Anti-E-tag抗体反应均为阳性的克隆扩大培养,进行菌液PCR,阳性者送去测序,测得序列经分析后共得到2条与驼源的VHH具有较高同源性并与GP5蛋白具有良好反应原性的序列,分别命名为Nb1、Nb2(图6)。分析可知,两株纳米抗体的FR2区均存在典型的亲水氨基酸。
2.9 重组质粒pcDNA3.1(-)-Nbs的构建
以所筛Nb1、Nb2所对应的菌液为模板,使用引物VHH-ZH(F)、VHH-ZH(R)对纳米抗体基因进行PCR扩增,分别得到片段大小约为340 bp的目的条带(图7A),将扩增出的纳米抗体基因克隆至真核表达载体pcDNA3.1(-)中,分别构建出重组质粒pcDNA3.1(-)-Nb1与pcDNA3.1(-)-Nb2,随后对重组质粒进行菌液PCR验证(图7B)与双酶切验证(图7C),并送去测序,结果显示,重组质粒pcDNA3.1(-)-Nbs构建成功。
2.10 重组质粒转染至Marc-145细胞
将构建好的重组质粒利用Lipofectamine3000瞬时转染至Marc-145细胞中,36 h后,收集细胞,分别检测细胞中Nb1与Nb2蛋白的表达情况。如图8A所示,转染后可于细胞中检测到Nb1与Nb2蛋白的表达。使用荧光定量PCR检测重组质粒转染细胞前后Nb1与Nb2 mRNA表达量的变化,如图8B与8C所示,两株纳米抗体的表达量于细胞转染后均显着增加,进一步验证了重组质粒pcDNA3.1(-)-Nbs转染成功。
2.11 细胞内表达的Nb1与Nb2对PRRSV复制的影响
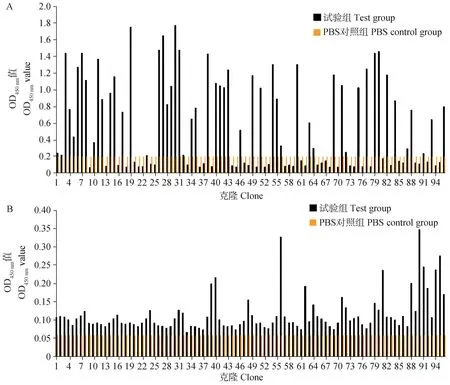
A. Anti-E-tag抗体(OD450 nm值为0~1.8;1~96. 试验组);B. Anti-M13抗体(OD450 nm值为0~0.35;1~96. 试验组) A. Anti-E-tag antibody (OD450 nm value is 0-1.8; 1-96. Test group); B. Anti-M13 antibody (OD450 nm value is 0-0.35; 1-96. Test group)图5 重组纳米抗体的间接ELISA检测Fig.5 Indirect ELISA detection of recombinant nanobodies
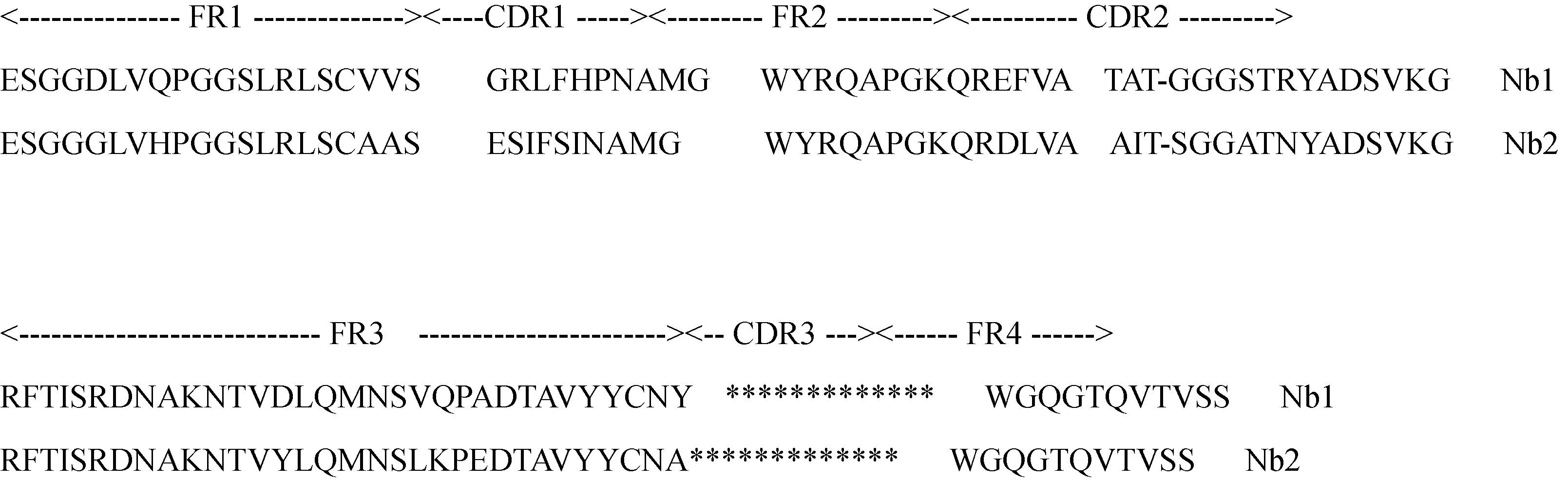
图6 Gp5特异性纳米抗体氨基酸序列Fig.6 Amino acid sequence of Gp5 specific nanobody
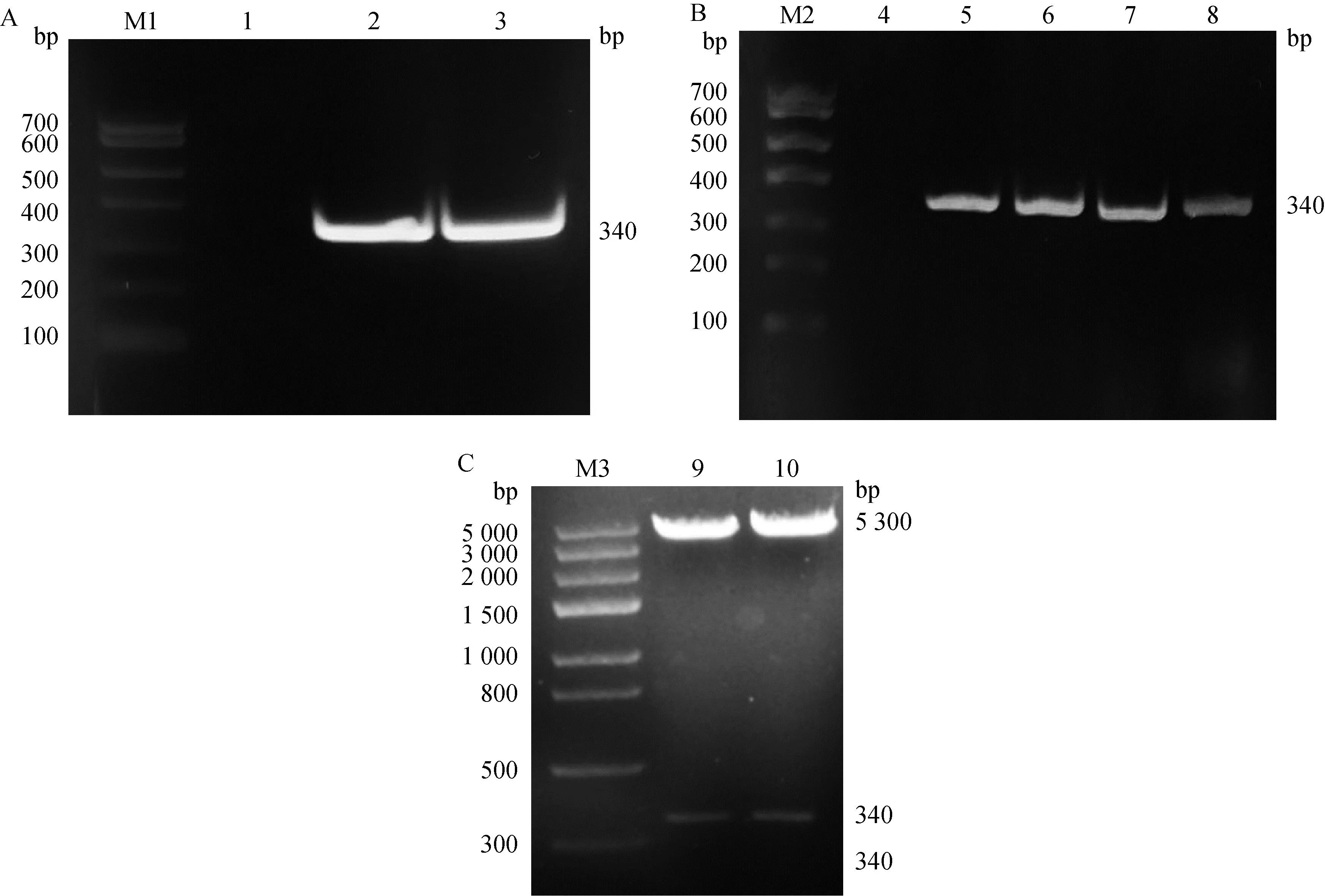
A. Nb1与Nb2基因的PCR扩增;B. 重组质粒pcDNA3.1(-)-Nb1与pcDNA3.1(-)-Nb2的PCR鉴定;C. 重组质粒pcDNA3.1(-)-Nb1与pcDNA3.1(-)-Nb2的双酶切鉴定;M1. DL700 DNA相对分子质量标准;1. 阴性对照;2. Nb1基因PCR扩增产物;3. Nb2基因PCR扩增产物;M2. DL700 DNA相对分子质量标准;4. 阴性对照;5~8. 菌液PCR扩增产物;M3. DL5000 DNA相对分子质量标准;9. pcDNA3.1(-)-Nb1重组质粒;10. pcDNA3.1(-)-Nb2重组质粒 A. PCR amplification of Nb1 and Nb2 gene; B. Identification of pcDNA3.1(-)-Nb1 and pcDNA3.1(-)-Nb2 by PCR; C. Identification of pcDNA3.1(-)-Nb1 and pcDNA3.1(-)-Nb2 by double enzyme digestion; M1. DNA marker DL700; 1. Negative control; 2. PCR amplification product of Nb1 gene; 3. PCR amplification product of Nb2 gene; M2. DNA marker DL700; 4. Negative control; 5-8. PCR amplification products of bacterial fluid; M3. DNA marker DL5000; 9. Recombinant pcDNA3.1(-)-Nb1; 10. Recombinant pcDNA3.1(-)-Nb2图7 重组质粒pcDNA3.1(-)-Nbs的构建Fig.7 Construction of pcDNA3.1(-)-Nbs recombinant plasmid
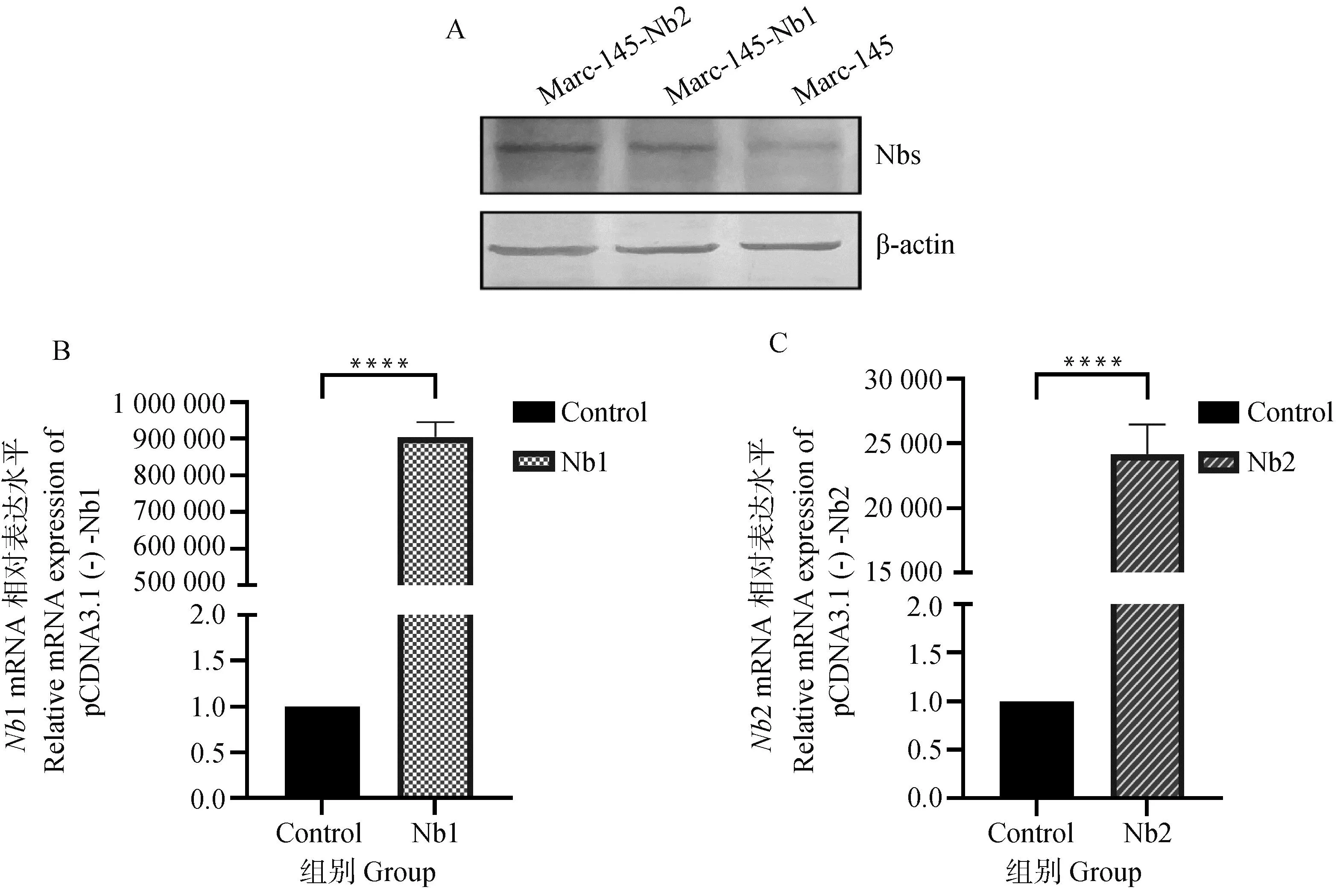
A. 重组质粒pcDNA3.1(-)-Nbs转染至Marc-145细胞的Western blot鉴定;B. 重组质粒pcDNA3.1(-)-Nb1转染至Marc-145细胞的荧光定量PCR鉴定;C. 重组质粒pcDNA3.1(-)-Nb2转染至Marc-145细胞的荧光定量PCR鉴定。与空白对照组相比,****.P<0.000 1 A. Westernblot identification of recombinant plasmid pcDNA3.1 (-)-Nbs transfected into Marc-145 cells; B. Fluorescence quantitative PCR identification of recombinant plasmid pcDNA3.1 (-)-Nb1 transfected into Marc-145 cells; C. Fluorescence quantitative PCR identification of recombinant plasmid pcDNA3.1 (-)-Nb2 transfected into Marc-145 cells. Compared with the Control group, ****. P<0.000 1图8 重组质粒pcDNA3.1(-)-Nbs转染至Marc-145细胞Fig.8 Recombinant plasmid pcDNA3.1 (-)-Nbs transfected into Marc-145 cells
将pcDNA3.1(-)-Nb1、pcDNA3.1(-)-Nb2分别转染至Marc-145细胞中,用1 MOI PRRSV感染各组细胞与空细胞对照组,分别收集0、24、36、48、60、72 h的细胞样品进行RT-qPCR检测PRRSVORF7基因 mRNA表达水平的变化。如图9A所示,病毒与细胞共孵育24 h内,Marc-145-Nb1细胞内的病毒载量与对照组无显着差异,而在36~72 h内,Marc-145-Nb1细胞内所含病毒量显着低于对照组,说明Nb1可以在细胞内抑制PRRSV的复制与转录。如图9B所示,在病毒与细胞孵育48 h内,Marc-145-Nb2细胞内所含病毒量与对照组无显着差异,而于60~72 h内,Marc-145-Nb2细胞内的病毒载量显着低于对照组,这说明Nb2亦具备抑制PRRSV复制的能力,但较于Nb1对病毒复制的抑制效力,Nb2抑制病毒复制的时间出现较晚,且随时间的推移其阻碍病毒复制的能力呈下降趋势,这些因素均说明Nb1较Nb2具备更强的阻碍病毒复制的能力。
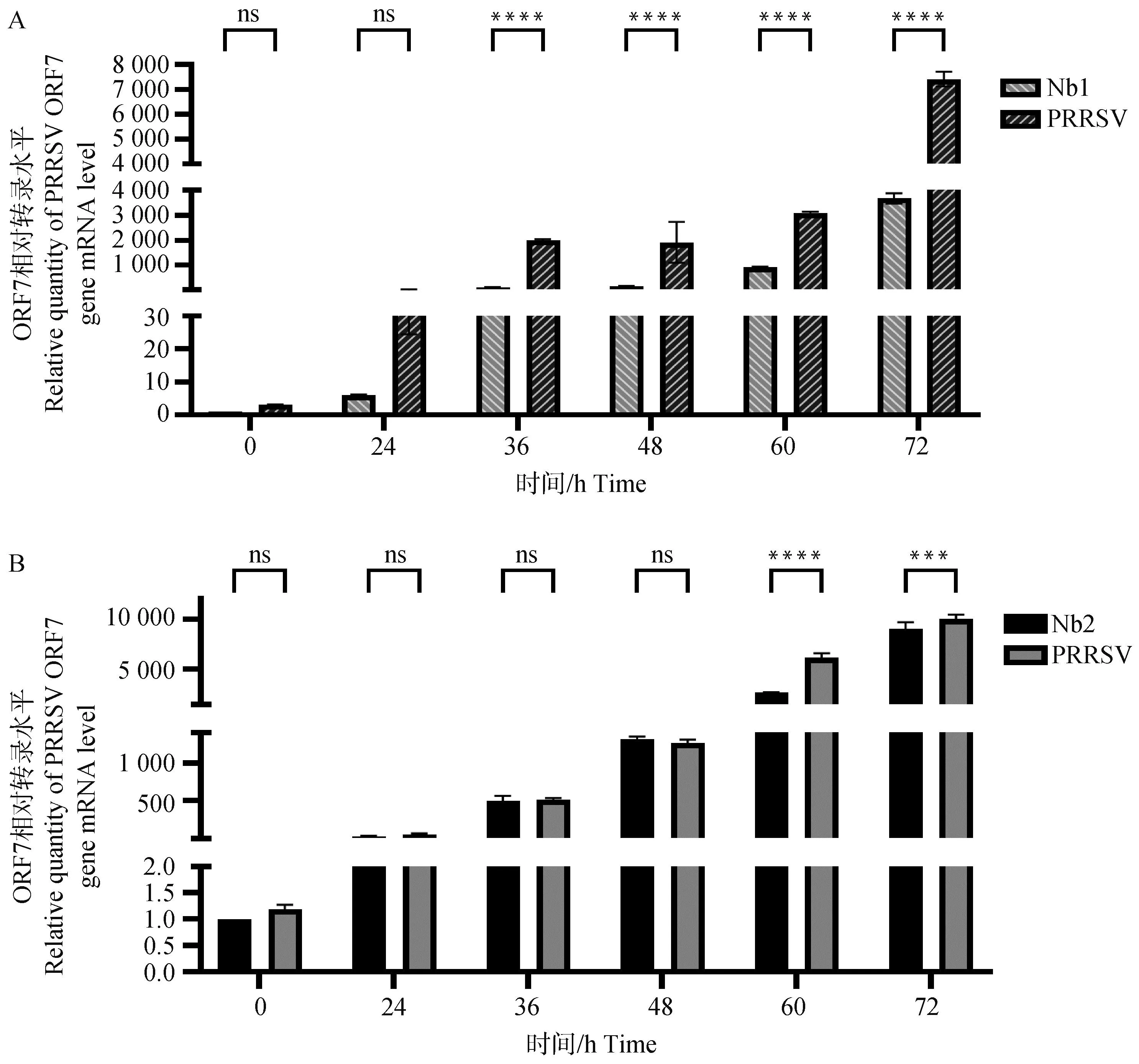
A. Marc-145-Nb1对PRRSV复制的影响;B. Marc-145-Nb2对PRRSV复制的影响。与PRRSV对照组相比,ns表示无显着性差异,***.P<0.001,****.P<0.000 1 A. The effect of Marc-145-Nb1 on PRRSV replication; B. The effect of Marc-145-Nb2 on PRRSV replication. Compared with the PRRSV control group, ns represents no significant difference, ***. P<0.001, ****. P<0.0001图9 细胞内表达的Nb1与Nb2对PRRSV复制的影响Fig.9 The effect of intracellular expression of Nb1 and Nb2 on PRRSV replication
3 讨 论
目前,猪繁殖与呼吸综合征(俗称蓝耳病)在全球仍呈不断传播与流行的趋势。在我国,该病已流行二十多年,PRRSV特别是高致病性PRRSV的变异株始终是严重威胁我国养猪业的最重要病原之一。其中PRRSV GP5蛋白氨基酸序列的高突变性可能给猪群的疫苗保护带来威胁,为蓝耳病的防控带来了不利影响[15],因此,对蓝耳病的诊断与治疗具有重大意义。GP5蛋白作为PRRSV中的重要结构蛋白,可以刺激机体发生T细胞的增殖反应,诱导细胞免疫产生[16]。Wang等[17]使用GP5蛋白作为抗原进行酶联免疫吸附试验,发现该蛋白具有较高特异性,是一种优异的诊断抗原。Du等[18]使用ORF5基因制备的疫苗免疫仔猪与小鼠,发现疫苗可以使宿主细胞的黏膜免疫有所增强,且能够显着提高仔猪与小鼠血清中IgG水平以及肠道内IgA水平。因此,GP5蛋白凭借其具备良好的免疫原性以及较强的中和活性,已成为蓝耳病相关抗体制备及疫苗研发的优秀候选蛋白。
纳米抗体作为一种较为新颖且亲和力强的抗原识别工具,可能因其凸起的对角以及灵活的CDR3环形成loop环,可与蛋白质空间结构上的裂隙与空腔进行结合,从而能识别隐蔽的病毒表位与酶的活性位点,这意味着纳米抗体有助于潜在生物靶标的预测以及新的药理靶点的发现[19]。与传统抗体相比,纳米抗体具备体积小、易于表达与改造等优势,其可以直接靶向作用于抗原表位,亦可以构建成纳米抗体融合蛋白或是多价纳米抗体以发挥治疗作用[20-21]。近年来,纳米抗体在医学领域抗体研究方面发挥着不可或缺的作用,且在疾病的诊断与治疗方面的研究持续推进[22]。刘红亮[13]针对PRRSV Nsp9蛋白筛选出的Nb6纳米抗体可以抑制多种PRRSV毒株在细胞中的增殖与复制。宋欢等[23]制备出PRRSV Nsp2蛋白纳米抗体,结果显示所筛纳米抗体具有高度特异性,可满足其作为诊断与治疗抗体的要求。
本研究首先利用原核表达系统对PRRSV-GP5蛋白进行表达,经镍柱亲和层析法纯化后免疫羊驼,三免14 d后羊驼体内抗体效价高达1∶819 200,后通过噬菌体展示技术构建GP5-VHH噬菌体抗体展示文库。噬菌体展示技术源自于将外源多肽于单链噬菌体表面上展示的相关研究[24],其原理为将编码外源蛋白的基因序列或外源多肽插入至编码噬菌体衣壳蛋白的基因中,从而使得外源蛋白或多肽在噬菌体的表面得以展示[25]。经鉴定,所获噬菌体文库的库容量达2.93×107CFU·mL-1,插入率达98%。后进行连续三轮“吸附-洗脱-富集”的特异性噬菌体淘选,结果显示,重组噬菌体得到了有效富集,且每轮的洗脱量与回收率均逐渐升高,最终获得2株不同氨基酸序列的纳米抗体,间接ELISA显示,所筛纳米抗体均与PRRSV-GP5蛋白具有良好的亲和力。随后将所获的2个Nb基因克隆至真核表达载体pcDNA3.1(-)中,利用脂质体瞬时转染入Marc-145细胞内,与PRRSV共孵育以探究两株纳米抗体于胞内对病毒复制与转录的影响,结果显示所筛的Nb1与Nb2均可在细胞内抑制PRRSV的复制,且Nb1对病毒复制的抑制效力要优于Nb2,这意味着Nb1可成为新型抗PRRSV药物的候选者。
4 结 论
成功表达与纯化PRRSV-GP5蛋白,免疫羊驼后,利用噬菌体展示技术构建出GP5-VHH噬菌体抗体展示文库,首次筛选出针对PRRSV-GP5蛋白的特异性纳米抗体,且该纳米抗体于细胞内展现出较好的抑制病毒复制的能力。研究结果为抗PRRSV新型药物的研发提供了新的思路与试验依据。



