邱梅玉,张雪梅,张 宁,刘明军
(新疆畜牧科学院生物技术研究所 农业农村部草食家畜遗传育种与繁殖重点实验室新疆动物生物技术重点实验室,乌鲁木齐 830026)
在过去的十年中,CRISPR/Cas9技术允许精确靶向基因组位点的修饰是具有变革性的。在真核生物中,当不存在修复模板时双链断裂(double strand break, DSB)可能引入错配碱基对的非同源末端连接修复(non homologous end joining, NHEJ)途径对断裂位点进行修复,这种修复会造成碱基插入或缺失,从而导致该基因编码蛋白的移码突变引起基因敲除;而当存在修复模板时,精确的同源重组修复(homologous recombination repair,HDR)途径对断裂位点进行修复实现基因敲入或点突变[1]。细胞倾向于利用 NHEJ 来修复 DSB,造成 HDR 的效率偏低,因此 CRISPR/Cas9 系统无法高效地诱导点突变等编辑类型[2]。此外,通常情况下 NHEJ 和 HDR 两种修复途径同时存在且相互竞争,而且 NHEJ 效率比 HDR 高。因此,大多数编辑结果通常会同时发生插入或删除(insertions and deletions, Indels)编辑[3]。
随着CRISPR/Cas9 技术的不断发展衍生出碱基编辑技术 (base editing) ,通过将失去切割能力的核酸酶与不同碱基脱氨基酶融合,构建胞嘧啶碱基编辑器 (cytosine base editor,CBE) 和腺嘌呤碱基编辑器 (adenine base editor,ABE)。这两类编辑器能够在不产生 DNA 双链断裂的前提下在基因靶位点完成 C>T (G>A) 或 A>G (T>C) 的替换,最终实现精准的碱基编辑。目前碱基编辑技术已经广泛应用于基因治疗、动物模型构建、精准动物育种和基因功能分析等领域[4]。然而,单碱基编辑器会在高效编辑单碱基的同时产生严重的脱靶突变,并且不能实现碱基之间的颠换(A-T 或 C-G)[5-6]。
引导编辑系统(prime editing,PE)通过对靶位点的“搜索”和“替换”,可以实现 12种碱基之间的任意颠换及小片段的精准插入和删除,克服CRISPR/Cas9 系统介导的 HDR 效率低和碱基编辑系统不能实现碱基颠换的弊端,具有明显的优势[7-8]。
1 引导编辑系统的原理及特点
引导编辑系统包括 Cas9 切割酶(Cas9 nickase,nCas9)、逆转录酶 (reverse transcriptase,RT)和 引导 RNA(prime editing guide RNA,pegRNA),其中 nCas9 与 RT 融合在一起。pegRNA 的 3′端又延伸出了一段 RNA 序列,包括引物结合位点(prime binding site,PBS)序列和含有目标编辑序列的RT 模板 (reverse transcriptase template,RT template)[9]。利用 nSpCas9 (H840A)和逆转录酶(moloney murine leukemia virus reverse transcriptase,M-MLV RT)融合构建引导编辑器(prime editor),由 pegRNA 中 guide RNA 部分引导在人细胞基因组靶位点附近形成编辑链上的单链切口,进而通过pegRNA中的PBS序列引导以含有目标编辑序列的逆转录模板将突变精确导入基因组中(图1)[10-11]。
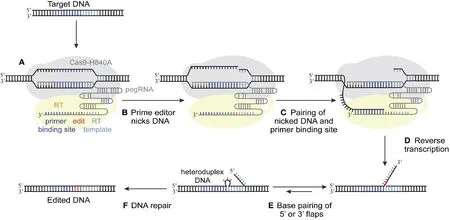
图1 引导编辑系统模式图(引自[11])Fig.1 Schematic Illustration of Prime Editing, as Proposed by [11]
相较于 CRISPR/Cas9 系统通过 HDR 途径引入点突变、插入或删除,引导编辑系统不依赖 DSB 避免产生过多的 indels 。碱基编辑系统作用过程中会引起邻近非靶点碱基编辑,而引导编辑系统通过逆转录模板产生目标突变避免indels的产生。对PE3 来说,虽然另一条 sgRNA 的引入造成indels 的增加,但 indels 的水平大多数在10%以下[7]。另外,CRISPR/Cas9 和碱基编辑系统均存在sgRNA 依赖性脱靶,碱基编辑系统还存在sgRNA 非依赖性脱靶和RNA 脱靶,而引导编辑系统在基因组靶位点上引入目标编辑过程中,需要发生 3 种核酸杂交:靶向区域 DNA 与 pegRNA 的 spacer 的杂交、靶向区域 DNA 与 pegRNA 的 PBS 序列的杂交、靶向区域 DNA 与逆转录产物的杂交,3 种杂交的存在决定引导编辑系统在理论上很难脱靶[12]。
2 引导编辑的编辑效率优化及衍生技术
引导编辑系统虽然优势明显,但其早期版本的编辑效率均较低,极大限制了其应用(表1)。目前,Liu 等[13]在 SpCas9 与 M-MLV 序列的 N 端和 C 端添加 c-Myc NLS 和 SV40 NLS,构成PE2*;用 SaCas9 和 SaCas9KKH 变体替换 SpCas9,构成 SaPE2*和 SaKKHPE2*编辑效率分别为5.6%~11.3%和1.3%~4.2%。Xu等[14]构建 PE-P2,利用自切割多肽 2A 串联表达引导编辑系统和筛选标记,强化sgRNA使水稻基因组中靶位点突变效率从 1.2%提高到 26%。同时发现在 RT 模板中引入同义错配碱基和 nCas9 的 C 端融合 M-MLV 均可提高引导编辑系统的编辑效率,结合使用时水稻、玉米原生质和人细胞中的平均编辑效率分别达到 24.3%、6.2%和 12.5%[15]。Lu 等[16]通过密码子优化和 CaM35 S 启动子优化提高PE在番茄中的编辑效率。
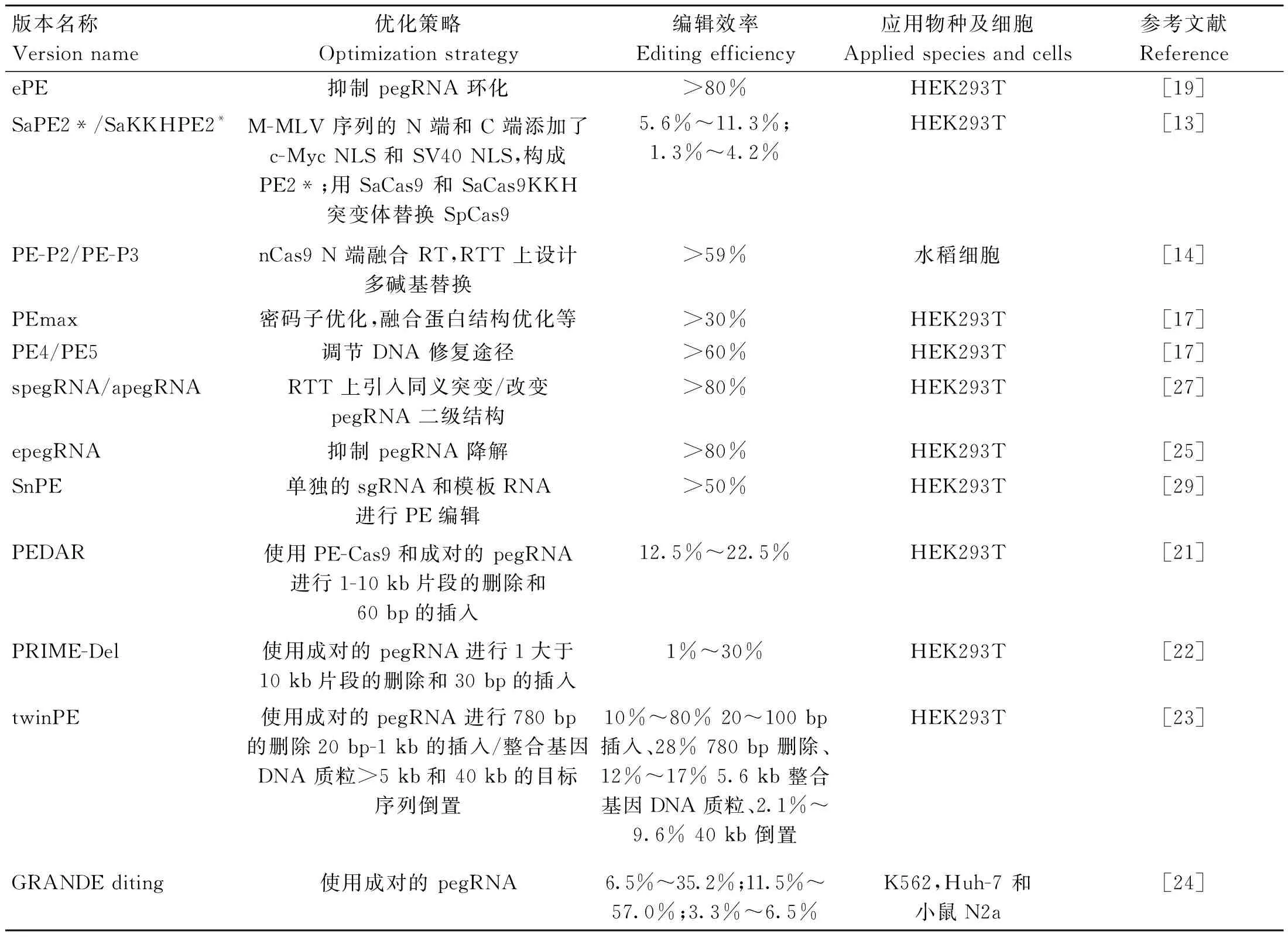
表1 引导编辑的衍生技术Table 1 Technologies derived from prime editing
对 nCas9-MMLV 融合蛋白多个方面进行优化(MMLV 的优化密码子、 Cas9n 突变体、Cas9n 与 MMLV 添加连接序列和增加 NLS 序列),构建出具有更高编辑效率的 PEmax[17]。
为了进一步提高引导编辑系统的编辑效率,Chen等[17]、Zhang等[18]、 Liu等[19]进行CRISPR 干扰(CRISPR interference,CRISPRi) 筛选,发现 DNA 错配修复(DNA mismatch repair,MMR)相关基因抑制引导编辑的效率,并增加 indels比例。Ferreira Da Silva 等[20]研究表明,通过消除 MMR,引导编辑系统的编辑效率可提高2~17 倍。Jiang 等[21]在引导编辑系统基础上研发 PE-Cas9-based deletion and repair (PEDAR),精确进行1~10 kb片段的删除和60bp的插入编辑效率为12.5%~22.5%,Choi等[22]设计PRIME-Del 编辑系统精确进行大于10 kb片段删除和30bp插入编辑效率为1%~30%。Anzalone 等[23]开发 twin prime editing (twinPE) 编辑系统使用成对的pegRNA 进行780 bp 的删除、20 bp~1 kb 的插入、整合基因DNA质粒>5 kb和 40 kb的目标序列倒置,相比PEDAR 和 PRIME-Del 会产生更少的indels。Wang 等[24]开发 GRANDE diting 系统,利用一对特殊设计的 pegRNA 在不同细胞中编辑实现长达 1 kb DNA 片段的靶向插入编辑效率分别为6.5%~35.2%、11.5%~57.0% 和3.3%~6.5%。
引导编辑系统发挥作用时pegRNA 的 3′端延伸未被融合蛋白包裹,容易被核酸酶降解。为解决这一问题,通过对 pegRNA 的 3′端添加 RNA 结构基序(G-quadruplex[25]、xrRNA[26]G-四链体[25]和 tevopreQ1(trimmed evopreQ1)[25]等)来增加 pegRNA 的稳定性,进一步提高引导编辑系统的编辑效率。添加 tevopreQ1 的epegRNA 与 MLH1dn 和 PEmax 协同作用可显着提高编辑效率,其中 PE4max 和 epegRNAs 在 Hela 细胞和 HEK293 T 细胞中的编辑效率分别提高72 倍和3.5 倍;PE5max 和 epegRNAs 在 Hela 和 HEK293 T 细胞中的编辑效率分别提高12 倍和 1.6 倍。Zhang 等[18]在pegRNA的3′端添加病毒抗核糖核酸酶外基序(xrRNA)提高其对降解的抗性,发现xrPE在给定的细胞类型中碱基替换、小缺失和插入的编辑效率分别提高3.1、4.5和2.5倍。Liu等[19]将20 bp Csy4识别位点与经典pegRNA的3′端融合,该位点自然存在于I-F型CRISPR/Cas9系统中形成发夹可抑制pegRNA 环化。Li 等[27]通过在pegRNA的逆转录模板的适当位置引入同义突变来开发spegRNA,将PE的平均编辑效率提高353倍。通过改变pegRNA的二级结构来开发apegRNA,使PE的平均编辑效率提高2.77倍。当spegRNA和apegRNA用于PE3和PE5系统时,sPE3、aPE3、sPE5和aPE5系统的编辑效率都显着提高。尽管在单个pegRNA中连接间隔区和模板序列可以确保它们在引导编辑过程中的相近性,但分离这些成分也可进行PE编辑。Liu 等[28]设计了一个分离的pegRNA系统,该系统由靶向PE的正常sgRNA、含有PBS、RT模板的线性或环状引物编辑模板RNA(petRNA)组成。petRNA还包含MS2发夹,通过结合融合的MS2外壳蛋白,促进其募集到RT。Feng 等[29]建立了一种类似的编辑系统称为Split pegRNA引导编辑(SnPE)。这两项研究都表明,用单独的sgRNA和模板RNA进行PE编辑是可行的。另外,PBS 和 RT 模板的长度与编辑效率没有绝对的对应关系,通常需要设计不同长度的 PBS 和RT 模板进行编辑尝试,前期相关研究中建议 PBS 和RT 模板的起始长度为 13 nt和 10~16 nt,但是当编辑位点的 G/C 含量超出 40%~60%时可适当延长或缩短 PBS 长度,并且 RT 模板与 sgRNA 骨架 3′端连接的第一个碱基不能是 C[7]。Lin 等[30]发现引导编辑系统在植物中 PBS 的Tm 值为 30 ℃时编辑效率最高,Tm 值过低或过高都会影响编辑效率,用双 pegRNA 将编辑效率提高 1.8~4.2 倍,并且没有产生额外的插入或缺失突变。
另外,通过piggyBac 载体[31-32]、腺病毒载体[33]递送引导编辑系统或将引导编辑系统构成一元载体[34]、 引导编辑系统分离成多个载体进行基因编辑[30]。引导编辑系统中加入HPT 抗性基因构建 pPE2、pPE3/3b,发现水稻中pPE2系统可以在不同的基因组位点诱导编辑,在转基因T0植物中pPE2产生的突变体发生频率为0~31.3%,这表明pPE2的效率在不同的基因组位点和不同结构下差异很大,并将HPT-ATG 回复突变加入到筛选过程中构建 surrogate PE2 系统,该系统能够富集编辑细胞提高编辑效率[35-37]。Oh 等[38]利用 SpCas9 的同系物 FnCas9 构建新的引导编辑系统,SpCas9 是在 PAM 上游 3~4 bp 之间对非靶向链进行切割,而 FnCas9 的切割位置在 PAM 上游 6~8 bp 之间,且FnCas9 和 SpCas9 识别同样的 NGG PAM,因此 FnCas9 的引导编辑系统具有更大的编辑窗口,扩大PE 的编辑范围。
3 引导编辑系统的脱靶效应
目前针对全基因组的特异性分析发现不会产生显着的单核苷酸水平脱靶及indels,由于逆转录酶介导的在引物序列之外的延伸,引导编辑可能会在目标位点引入小的插入,但其脱靶率远低于 CRISPR/Cas9 和 BE 编辑系统[39]。Zong 等[40]测试engineered plant prime editor (ePPE)对pegRNA中错配的耐受性,包括在水稻原生质体中的间隔区或引物结合位点和间隔区中的错配,ePPE在内源性位点的脱靶效应包括11个pegRNA的间隔区中的1~3个错配,共导致29个脱靶位点。深度测序显示除了OsCDC48-T1(OT-22)外ePPE和PPE在所有检测位点都表现出非常低的引导编辑脱靶效率,与PPE相比ePPE表现出更高的诱导编辑效率。Nelson等[41]测试每个靶向位点的前4个位置的indel产生和核苷酸变化,并比较PE3处理后epegRNAs和未修饰的pegRNAs之间的脱靶编辑,发现在检测位点epegRNAs和pegRNAs均表现出≤0.1%的引导编辑脱靶和indels产生,表明epegRNAs和pegRNAs表现出相似的脱靶编辑水平。Gao等[42]进行基因组和转录组测序分析,发现在人类细胞中引导编辑不会产生 gRNA 非依赖性 DNA 和 RNA 脱靶。Geurts等[43]利用全基因组测序检测引导编辑修复类器官的脱靶效应,发现在靶位点产生不同的编辑效率和突变,并强调引导编辑系统在建模致癌突变方面的广泛适用性。Habib等[44]比较引导编辑和碱基编辑以纠正来自α1-抗胰蛋白酶(A1AT)缺乏症患者诱导多能干细胞中的疾病相关突变,发现引导编辑的逆转录酶结构域不会导致基因组中gRNA非依赖性脱靶突变。总之,引导编辑系统相比 CRIPSR/Cas9 和碱基编辑系统具有更高的特异性,也更加安全,但仍需进一步降低其脱靶风险。
4 引导编辑系统的应用
由于引导编辑系统强大的编辑功能及普遍性,已迅速应用在各物种及各个研究领域中,本文将概述其中的一些进展(表2)。
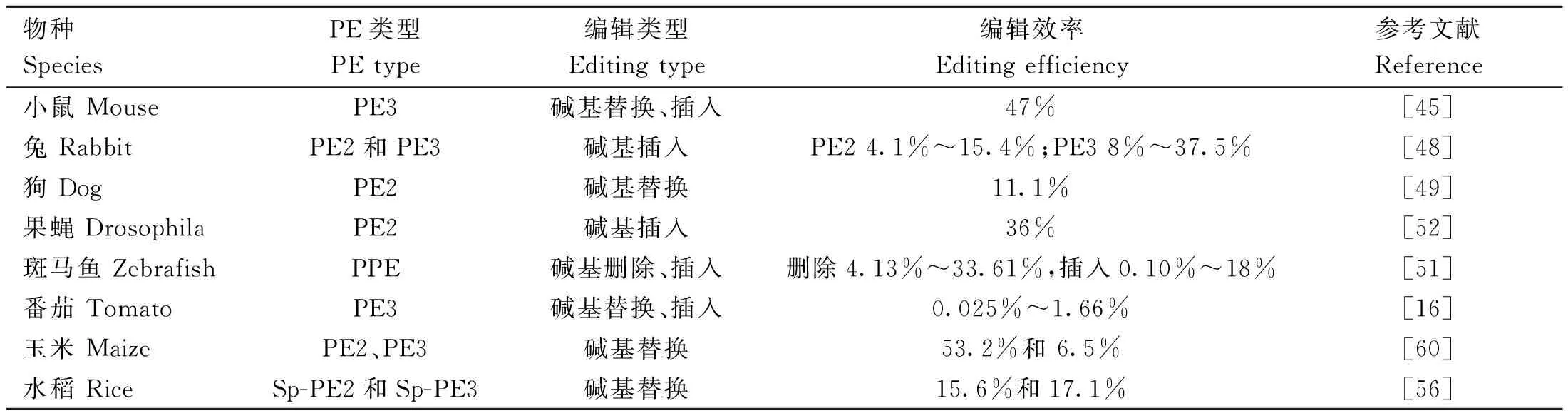
表2 引导编辑系统的应用Table 2 The applications of prime editing system
4.1 引导编辑系统在动物中的应用
Park等[45]利用引导编辑系统完成编辑频率高达47%的Igf2突变小鼠,并观察到该性状可在种系传递、无脱靶效应。Gao等[46]比较HDR和PE2在小鼠中对CArG box转录因子结合位点的编辑作用,对创制小鼠的分析发现成功编辑HDR(20/37)的动物数量大约是PE2(12/47)的两倍,但HDR创制的小鼠存在不同位置上的indels产生。研究表明使用PE腺病毒修复苯丙酮尿症小鼠模型中的Pah F263 S突变[47]。Qian等[48]利用引导编辑系统建立TSD兔模型,并对HEXA中的TATC兔模型中肌无力、共济失调和精神障碍的典型表型进行了表征,发现PE2和PE3编辑效率分别为4.1%~15.4%和8%~37.5%。Kim等[49]确定引导编辑在犬中的可行性并利用其生产犬髋关节发育不良相关基因矫正犬。Liu等[50]通过显微注射将PE3 mRNA和靶向Hoxd13(G/C和G/T)位点的pegRNA注射到小鼠胚胎中,编辑效率分别在27%和10.5%,indel效率在0%~0.3%。Petri等[51]在斑马鱼胚胎中使用纯化核糖核蛋白复合物(RNP)PE编辑技术引入所需的突变,编辑后发现精准删除和插入编辑效率分别为4.13%~33.61%和0.10%~18%并且形成较少的副产物。Bosch等[52]通过杂交在生殖细胞中表达gRNA和PE2蛋白的转基因果蝇产生引导编辑的黑腹果蝇。Atsuta 等[53]通过PE与转座子介导的基因组整合、药物筛选和单细胞培养方法相结合,产生引导编辑的鸡成纤维细胞和原始生殖细胞(PGCs)。Aida等[54]利用引导编辑技术在小鼠胚胎中引入10种靶向修饰包括替换、删除和插入,表明在哺乳动物受精卵中引导编辑具有高效性。
4.2 引导编辑系统在植物中的应用
在单子叶植物引导编辑系统中,研究表明PEs可以精确编辑水稻原生质体和小麦的基因组,其效率通常低于10%[38,55]。Hua等[56]在水稻中构建包含CaMV 35 S启动子驱动的非活性EGFP序列的Sp-PE2和Sp-PE3,通过后期筛选发现Sp-PE2和Sp-PE3水稻的编辑效率分别为15.6%和17.1%。Lin 等[57]通过密码子、启动子和编辑条件优化,使引导编辑系统能够在水稻和小麦原生质体中进行点突变、插入和缺失,获得再生的引导编辑水稻植株编辑效率高达21.8%。Xu等[58]利用引导编辑系统对水稻植株中的OsACC1进行突变,并在筛选鉴定出抗除草剂变异植株。引导编辑系统在双子叶植物中也表现出了活性,这些植物比单子叶植物更难转化。Lu等[16]通过优化引导编辑系统发现可介导二倍体番茄中0.025%~1.66%的基因编辑。Biswas等[59]在水稻、花生、鹰嘴豆和豇豆原生质体中利用引导编辑系统编辑了一个突变的GFP,在水稻中,双pegRNA的编辑效率是含单pegRNA的16倍。在用双pegRNA转化后,花生、鹰嘴豆和豇豆也获得经过编辑的突变GFP原生质体,尽管编辑效率比水稻低得多从0.2%~0.5%不等。Jiang等[60]利用引导编辑系统编辑玉米发现在ZmALS1和ZmALS2中分别有53.2%(33/62)和6.5%(4/62)的转基因系携带S621I和W542 L突变;在两个ALS基因中分别有4.8%(3/62)和4.8%(4/62)的品系携带纯合S621I突变和S621I/W542 L双突变。
4.3 引导编辑系统在基因治疗中的应用
基因治疗是基因编辑工具的一项重要应用,使用基因编辑工具直接修复致病突变有望治愈人类遗疾病[61]。基因治疗方法通常不能恢复自然条件下的基因调控,不能随着时间的推移而一直表达且递送大片段的基因,而治疗遗传疾病的理想方法是永久纠正基因组原位置的致病突变。引导编辑作为纠正人类基因组致病突变的治疗工具具有明显的优势,原则上可以纠正高达89%的与人类疾病相关的已知遗传变异。
在培养的细胞系中,引导编辑已被证明可以直接纠正HEXA中导致Tay-Sachs的4 bp插入,导致镰状细胞病的HBB E6V突变及CDKL5缺乏障碍的单碱基缺失[17]。在人类类器官模型中,引导编辑能够修复与DGAT1缺乏、威尔逊病和囊性纤维化相关的突变[46,62]。PEs可以通过精确删除SMN中的内含子剪接沉默来拯救脊髓性肌萎缩的人iPSC模型中全长SMN的表达[63]。Chemello 等[64]利用引导编辑技术插入2 bp,在Duchenne肌营养不良的人iPSC模型中恢复DMD阅读框。Geurts等[43]对囊性纤维化CFTR-F508del突变进行了功能修复,并将引导编辑与CRISPR/Cas9介导的CFTR-R785*突变的同源性定向修复和腺嘌呤碱基编辑进行比较,发现引导编辑修复类器官的全基因组测序结果中未检测到脱靶效应。Jang等[65]通过玻璃体内或视网膜下注射DNMT1和ATP78,发现引导编辑效率可达约1.8%的点突变。当来自隐性营养不良大疱性表皮松解症(RDEB)患者的COL7A1突变成纤维细胞时,利用引导编辑矫正成纤维细胞移植到免疫缺陷小鼠中获得RDEB模型[66]。Li 等[67]开发的一种矢量化引导编辑系统可在体内S镰状细胞病(SCD)小鼠模型(CD46/Townes小鼠)中直接修复造血干细胞(HSC)中的SCD突变,43%的HbS被HbA取代从而大大减轻SCD表型。
5 引导编辑系统设计工具
与长度只有20 nt的传统gRNA的基因敲除和碱基编辑相比,PE存在更复杂的pegRNA结构。虽然手工设计pegRNA是可行的,但也需要仔细考虑pegRNA的设计规则(包括原间隔子位置、有利的PBS和RT模板以及链方向)。然而,这使得过程复杂容易出错并且对大规模的设计存在限制。
目前已经开发一些在线软件,帮助设计合适的pegRNA和nick sgRNAs(ngRNA)(表3)。Hsu等[68]开发PrimeDesign用于 pegRNA 的设计(SpCas9)并利用此工具构建全面和可搜索的数据库—PrimeVar,该数据库包含超过68 500 种人类致病性遗传变异的模拟和纠正的 pegRNA 和ngRNA组合。Chow 等[69]开发pegFinder进行PE2和 PE3 pegRNA设计(支持SpCas9,SpCas9-NG,Cas9-SpRY),还给出不同长度的PBS 序列和RT 模板供科研人员选择。Anderson等[70]设计pegIT同时设计用于靶基因PCR扩增的引物,并且在软件中也报告pegRNA和ngRNA(SpCas9,SpCas9-NG,CjCas9,SaCas9,SaCas9-KKH)间隔区的潜在脱靶位点。Hwang等[71]向引导编辑系统应用者提供两个免费的网站 PE-Designer和 PE-Analyzer。PE-Analyzer是PE结果分析的专用工具接受高通量测序数据,在表中总结突变相关信息并提供交互式图表。PE-Designer主要是用JavaScript编写可进行 pegRNA(SpCas9,SpCas9-NG,SpCas9-VRER,SpCas9-VQR,Cas9-SpRY ) 的设计,包括目标序列潜在的靶位点、pegRNA 延伸序列及ngRNA的序列。Lin 等[30]基于PBS 序列设计和双pegRNA (NGG-pegRNA 和 CCN-pegRNA)的策略开发了植物 pegRNA ,为使用者提供包括PAM 序列、spacer 的 GC 含量、 引导编辑窗口、PBS 长度、推荐的 PBS的Tm 值和RT 模板同源臂长度等一系列参数选择。Standage-Beier等[72]开发PINE-CONE用于pegRNA的高通量设计。Siegner等[73]开发PnB Designer用于PE的pegRNA和BE(SpCas9)的gRNA的设计。Li等[74]开发基于机器学习的程序Easy-Prime,用于PE的pegRNA(SpCas9)的设计提高编辑效率。

表3 pegRNA 设计软件Table 3 Software for pegRNA designing
6 问题与展望
PE技术的快速发展,极大地提高对真核细胞基因组进行精确修饰的能力。与CBE和ABE产生的编辑效率分别为50%和40.87%~64.22%相比,引导编辑系统的编辑效率相对较低,目前研究均基于改造PE蛋白及pegRNA结构[75-76]。与其他基因编辑技术相比,引导编辑技术具有多能性、产物纯度和目标序列的特异性,推动了许多创新性研究,对于动植物精准育种和微突变引起的疾病治疗等方面有很大的应用潜力。
引导编辑系统已在多个物种中进行成功编辑,有广泛的普适性,但仍存在的问题限制了其在农业、畜牧业、基础生物医学研究和临床转化中的广泛应用。与Cas9相比PE可诱导较低的脱靶编辑,但当使用ngRNA时虽然提升了编辑效率同时也造成了更多的indels。PE编辑通常很难在没有重组酶的帮助下有效地插入或替换超过100 bp的DNA[77]。PE编辑器M-MLV RT部分中的RNase H结构域可以与蛋白质翻译终止因子eRF相互作用,促进终止密码子的通读,当完整的PE2被转染到细胞中时,可能会造成整体蛋白的翻译效率降低[78]。此外,通过消除MMR提升PE4和PE5编辑效率,但额外添加的MLH1dn使其尺寸达到约8.7 kb,限制了AAV包装和体内传递[17]。目前,有许多pegRNA设计工具可用,但没有一个提供排名评分来告知用户哪些是强烈推荐的pegRNA。因此,需要建立一个更全面、更准确的计算模型,以减少密集的pegRNA的构建和筛选过程。总之,引导编辑系统在动植物基因功能研究和育种方面有非常广阔的前景,并且该技术有可能成为一种新的基因治疗形式,以安全、高靶向的方式插入治疗基因替代突变或缺失的基因。



