王亚茹 熊小婷 夏泽敏 黄日林 谭焯针 佘文勋
【摘 要】建立了一种牙膏中蒙花苷的高效液相色谱串联质谱(HPLC-MS/MS)检测方法。样品经纯水充分分散后,经乙腈超声提取,采用HPLC-MS/MS多反应监测模式定性定量分析。对液相色谱条件及质谱条件进行优化,结果表明,在优化的条件下,蒙花苷在5.0~200.0μg/L质量浓度范围内呈良好线性相关性,相关系数(R2)为0.9987;检出限和定量限分别为0.015μg/g和0.05μg/g;选取低、中、高三个浓度5.0μg/L、50.0μg/L、100.0μg/L进行加标回收实验,平均回收率为91.4~95.0%,相对标准偏差<3.70%。本方法简单易行、灵敏可靠,可应用于实际牙膏样品中蒙花苷的测定。
【关键词】高效液相色谱串联质谱;蒙花苷;牙膏;检测
中图分类号: O657.63;TS264.2 文献标识码: A文章编号: 2095-2457(2019)31-0187-002
DOI:10.19694/j.cnki.issn2095-2457.2019.31.090
野菊花为多年生草本植物野菊的头状花序,外形近似菊花,具有治疗疔疮痈肿、咽喉肿痛、风火赤眼、头晕目眩等症状,同时可以作为降血压和血糖的辅助疗法,在牙膏中添加野菊花提取物具有抑菌消炎、抗不良刺激的作用。蒙花苷是野菊花的主要药效成分,也是我国2010和2015版药典中明确规定的野菊花药材的质控指标[1]。近年来,一些植物提取物被商家以功效成分添加到牙膏中,以此对外宣称为功效型牙膏,野菊花提取物蒙花苷便是其中的典型代表。行业标准QB/T 2966-2014《功效型牙膏》中规定,功效性牙膏中的功效成分应进行定性定量检测[2],然而,目前尚无针对功效型牙膏中蒙花苷含量检测的方法标准。因此,开展对牙膏中功效成分蒙花苷的检测研究,对于帮助生产企业实现产品质量监控、保护消费者权益具有重要意义。
目前蒙花苷的检测研究报道多针对药材及中药制剂[3-7],报道的检测方法主要有高效液相色谱法(HPLC)和超高效液相色谱法(UPLC)。对于牙膏中蒙花苷的检测研究,刘广源等[8]曾报道了一种采用HPLC测定牙膏中蒙花苷含量的方法。然而,在实际检测中发现,由于牙膏基质成分复杂,采用HPLC分析时会存在杂质干扰严重或容易产生假阳性结果的情况。牙膏中蒙花苷添加含量一般为数μg/g,采用HPLC分析时需更大样品量,同时杂质干扰严重,为解决HPLC的不足,本文采用HPLC-MS/MS法建立了牙膏中蒙花苷含量检测方法。HPLC-MS/MS法集HPLC和MS/MS的优点于一体,既能实现快速分析,又能准确定性,同时具有更高的灵敏度。本文所建方法,样品经水和乙腈提取后,上机分析仅需8min,大大提高工作效率,且有着较高的灵敏度,目标物在样品中的检出浓度可达0.015μg/g。
1 试验
1.1 主要仪器与试剂
主要仪器:Agilent 1290Ⅱ-6495B高效液相色谱串联质谱仪,美国安捷伦有限公司;色谱柱:Agilent Poroshell 120 SB-C18(2.1×100mm,2.7μm),美国安捷伦有限公司;超声波清洗仪,上海科导超声仪器有限公司;高速离心机,曦玛离心机(扬州)有限公司;万分之一分析天平、十万分之一分析天平,赛多利斯科学仪器(北京)有限公司;纯水仪,美国Merck millipore公司。
主要试剂:蒙花苷标准品,纯度99.2%,上海安谱科学仪器有限公司;乙腈、甲醇,色谱纯,美国默克公司;甲酸、乙酸铵,优级纯,上海安谱科学仪器有限公司;超纯水,由纯水仪制备。
1.2 标准储备溶液的配制
精密称取0.0100g的蒙花苷标准品于10mL棕色容量瓶中,用二甲基亚砜溶解并定容至刻度,配置成1000mg/L标准储备液,4℃避光保存。
1.3 标准工作溶液的配制
准确移取适量体积的蒙花苷标准储备溶液(1.2)于10mL棕色容量瓶中,用乙腈配制成浓度为10.0mg/L的标准中间溶液,临用前用乙腈稀释成浓度为5.0、10.0、20.0、50.0、100.0、150.0、200.0μg/L标准工作溶液。
1.4 样品前处理
准确称取牙膏样品1.0g(精确至0.001g)于10mL具塞棕色比色管中,加入2mL超纯水,涡旋30S,再加乙腈定容至刻度,涡旋30s,超声20min,涡旋混匀,静置10min,取上清液过0.22μm有机滤膜,待测。
1.5 仪器条件
色谱条件:色谱柱:Proshell 120 SB-C18(2.1*100mm,2.7μm);流速:0.3mL/min;流动相:A:0.1%甲酸水溶液,B:乙腈;洗脱程序:梯度洗脱,0~2min,流动相A比例为90%,2~3min,流动相A由90%变为30%,3~6min,流动相A 30%保持不变,6.1~8min,流动相A比例为90%;进样体积:3μL。
质谱条件:离子源:ESI源;检测模式:多反应监测(MRM)模式;扫描模式:正离子模式;干燥气温度: 325℃,流速:6.0 L/min;鞘气温度:300℃,流速:11.0L/min,定量离子对:593.3→285.3,碰撞能量:29eV;定性离子对:593.3→447.4,碰撞能量:17 eV;碎裂电压380 V。
2 结果与讨论
2.1 质谱参数的优化
对浓度为500.0μg/L的蒙花苷标准溶液在电喷雾离子源下分别进行正离子和负离子全扫描分析,结果显示,正离子模式下获得分子离子峰的响应较佳,因此选择正离子模式下进行分析。采用子离子扫描方式进行二级质谱分析,选取丰度较强、干扰较小的两个特征碎片离子分别作为定量离子和定性离子。针对所选定量离子和定性离子对碰撞能量、干燥气温度和流速等条件进行进一步优化,确定最终的质谱分析条件。
2.2 流动相的选择
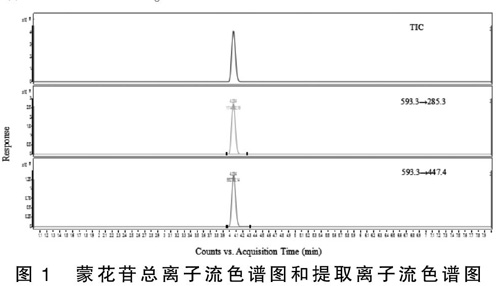
牙膏样品基质较为复杂,为了更好地降低基质对分析结果的影响,色谱分离采用了梯度洗脱的方式进行。对比考察了0.1%甲酸水溶液+甲醇、0.1%甲酸水溶液+乙腈、0.1%甲酸水溶液+0.1%甲酸乙腈溶液三组流动相的分析效果,结果显示,三组流动相都能实现良好分离,但采用乙腈作为流动相时,蒙花苷的响应较好。因此,最终选取0.1%甲酸水溶液+乙腈作为流动相。优化后的的总离子流色谱图和提取离子流色谱图如图1所示。
图1 蒙花苷总离子流色谱图和提取离子流色谱图
2.3 线性、检出限和定量限
将系列标准工作溶液(1.3)按照1.5的分析条件进行测定,以定量离子对色谱峰面积(Y)为纵坐标,待测组分蒙花苷的质量浓度X(μg/L)为横坐标,绘制校准工作曲线。结果表明,蒙花苷在5.0~200.0μg/L范围内呈良好的线性相关性,线性回归方程为Y=500.66X+174.70,相关系数(R2)为0.9987。方法检出限(S/N=3)为0.015μg/g,方法定量限(S/N=10)为0.05μg/g。
2.4 方法的回收率和精密度
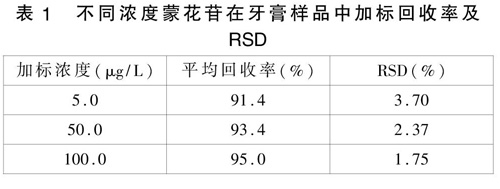
选择未检出蒙花苷的牙膏样品作为加标基质,分别加入一定体积的标准溶液,得到加标浓度分别为5.0、50.0、100.0μg/L的低、中、高3个加标浓度水平,每个浓度水平进行6次平行测定,结果见表1。结果显示,三个浓度水平的平均回收率在91.4~95.0%之间,相对标准偏差(RSD)均小于3.70%。结果说明本方法准确性高、重现性好,可以满足日常牙膏中蒙花苷的测定要求。
表1 不同浓度蒙花苷在牙膏样品中加标回收率及RSD
2.5 实际样品的检测
从超市随机购买5批外包装标识含有野菊花提取成分的牙膏样品,采用本文所建方法进行测定。结果显示5批样品均未检出蒙花苷。
3 结论
本文建立了一种牙膏中野菊提取成分蒙花苷的高效液相色谱串联质谱检测方法。方法简单易行、灵敏可靠,可应用于实际牙膏样品中蒙花苷的测定。对5批标识含有野菊花提取成分的牙膏样品进行测定,结果均未检出蒙花苷。
【参考文献】
[1]王维娜,余琦.高速逆流色谱分离纯化野菊花中蒙花苷和木犀草素[J].科技创新导报,2016,13(1):159-160.
[2]QB/T 2966-2014,功效型牙膏[S].
[3]张旭,胡晓梅,罗霄,等.HPLC测定甘松中的蒙花苷[J].华西药学杂志,2007,22(6):690-692.
[4]朱露,雷鹏,刘海涛,等.HPLC同时测定密蒙花中毛蕊花苷、蒙花苷的含量[J].中国实验方剂学杂志,2014,20(13):76-79.
[5]韩永成,龚海燕,刘伟,等.UPLC同时测定野菊花中蒙花苷和绿原酸含量[J].中国实验方剂学杂志,2013,19(20):88-90.
[6]黄兰芷,刘毅,蔡艳华.RP-HPLC对野菊花药材中蒙花苷和木犀草素的含量测定[J].中成药,2009(8).
[7]唐维宏,韦建华,马云婷.HPLC测定密蒙花配方颗粒中蒙花苷含量[J].中国实验方剂学杂志,2013(20).
[8]刘广源,张琥,费振玉.高效液相色谱法测定牙膏中蒙花苷的含量[C].中国口腔护理用品工业学术研讨会,2014.



