周建浩,王东方,刘 影,王淑娟,马震原,谢彩华,赵雪丽,杨海波,冯桂丹, 康台生,胡煜锋,李博文,闫若潜*
(1.河南农业大学动物医学院,郑州 450046;2.河南省动物疫病预防控制中心/河南省重大动物疫病监测预警 及防控重点实验室,郑州 450008;3.河南科技大学动物科技学院,洛阳 471000; 4.上海市动物疫病预防控制中心,上海 201103)
猪流行性腹泻(porcine epidemic diarrhea,PED)是由猪流行性腹泻病毒引起的一种急性、高度接触性的肠道传染病,以急性腹泻、呕吐、脱水为主要特征[1]。PED是当今养猪业除非洲猪瘟以外危害最为严重的腹泻疫病,该病主要对10日龄内仔猪危害最为严重,发病率可达100%;致死率为80%,最高可达100%[2],我国将其列为二类动物疫病。PEDV是α冠状病毒属(Alphacoronavirus)的成员,直径为90~190 nm的RNA病毒,该基因组长约28 kb,包含2个复制酶蛋白:ORF1a和ORF1b;4个结构蛋白:S、E、M、N;1个非结构蛋白:ORF3。我国当前PEDV毒株主要分为经典毒株(G1)和变异毒株(G2)两大群,经典毒株以CV777为代表,分为G1a和G1b两个亚群;变异毒株分为G2a、G2b和G2c三个亚群,以G2a(如AH2012、JS-HZ2012等)最为流行[3]。2010年11月以来,由PEDV变异毒株(G2群)引起的猪群腹泻性疫病可感染各个阶段的猪,尤其对哺乳仔猪危害最严重,原有的以CV777毒株代表的经典毒株疫苗对变异株感染保护效力差。PEDV经典毒株和变异毒株基因组中的M和N基因高度保守,因此本研究建立的ddPCR方法引物/探针的选择根据M基因保守区域进行设计,以实现可对PEDV经典毒株和变异毒株同时检测的目的。
数字PCR(digital PCR,dPCR)是继普通PCR、荧光定量PCR之后出现的第三代聚合酶链式反应(polymerase chain reaction)技术[4-5]。目前数字PCR技术主要分为微滴式数字PCR(droplet digital PCR, ddPCR)和芯片式数字PCR[6](chip digital PCR,cdPCR)。dPCR与荧光定量PCR相比较,dPCR不受扩增效率的影响,无需依赖标准曲线和参照样本[7],极大提高了低拷贝样品的检测灵敏度,因而定量结果更加准确[8]。dPCR方法灵敏度高、特异性好,实现了真正意义的绝对定量[9],更有利于低病毒含量样品的检测进而推动PEDV核酸标准物质的研制。
本研究将根据PEDV的M基因的保守区域设计的引物对/探针,建立特异性强、敏感性高、重复性好的ddPCR检测方法,为临床上PEDV感染的早期检测诊断、PEDV核酸标准物质的研制等提供技术手段。
1 材料与方法
1.1 仪器设备及主要试剂
全自动核酸提取仪、商品化病毒基因组DNA/RNA核酸试剂盒为西安天隆公司产品;ABI 7500荧光定量PCR仪为美国ABI公司产品;微滴式数字PCR仪,2×PerfeCTa® qPCR ToughMix® UNG、2×qScriptTMXLT 1-Step RT-qPCR ToughMix®、Fluorescein sodium Salt等试剂为法国Stilla Technologies公司产品;PrimeScriptTMRT Master Mix、Premix ExTaqTM等均为宝生物工程(大连)有限公司产品。
1.2 样品来源
猪流行性腹泻病毒(PEDV)、猪丁型冠状病毒、猪伪狂犬病病毒、猪传染性胃肠炎病毒、猪急性腹泻综合征冠状病毒、猪轮状病毒、猪细小病毒、猪瘟病毒、猪繁殖与呼吸综合征病毒、猪圆环病毒2型等10种病毒核酸样品由河南省动物疫病防控中心实验室提供;口蹄疫病毒O型、A型国家二级标准物质由河南省动物疫病预防控制中心实验室提供。疑似PEDV感染的肠内容物、小肠组织、粪便等150份样品由河南省动物疫病预防控制中心实验室保存。
1.3 ddPCR引物和探针的设计及合成
根据GenBank登录的PEDV的基因序列,选取其M基因(MK862249.1)序列保守区,利用Primer Express 3.0软件设计特异性引物和探针,引物对/探针序列为F:5′-GGCGCAGGACACATTCTTG-3′;R:5′-AAGTAGTGAGAAGCGCGTCTGTT-3′;P:FAMTGGTCTTTCAATCCTG-MGB。引物及探针均由宝生物工程(大连)有限公司合成。
1.4 ddPCR反应程序的优化
1.4.1 ddPCR预混酶的选择 使用自行建立的FQ-PCR引物对/探针。将经FQ-PCR检测定量后的PEDV核酸稀释至终浓度1.0×104copies·μL-1作为一步法ddPCR模板,将其核酸RNA反转录为cDNA作为两步法检测模板(以下简称‘模板’)。在一步法和两步法ddPCR supermix for Probes 体系分别加入2×PerfeCTa® qPCR ToughMix® UNG、2×qScriptTM XLT 1-Step RT-qPCR ToughMix®进行扩增。根据两种预混酶扩增结果,选择合适的预混酶。
1.4.2 ddPCR引物对/探针浓度以及退火温度的优化 以PEDV核酸cDNA为模板,ddPCR引物的浓度为0.3~0.9 μmol·L-1、探针浓度0.1~0.3 μmol·L-1,采用矩阵法筛选最佳的引物对/探针组合浓度。退火温度设置52~62 ℃。反应体系:2×PerfeCTa® qPCR ToughMix® UNG,Fluorescein sodium Salt 2.5 μL,模板3 μL,用无菌蒸馏水补齐至25 μL。ddPCR扩增条件:45 ℃ 5 min,95 ℃ 3 min;95 ℃ 15 s,50~62 ℃ 45 s,共45个循环。通过分析微滴分布状态、拷贝数、检测结果等确定最佳的引物对/探针浓度和最佳退火温度。
1.5 ddPCR检测方法与行业标准FQ-PCR方法敏感性比较试验
对PEDV核酸cDNA模板进行10倍梯度稀释,浓度为1.0×104~1.0×10-1copies·μL-1;采用中华人民共和国出入境检验检疫行业标准实时荧光RT-PCR检测方法[10](以下简称:行标FQ-PCR方法)和本研究建立的ddPCR方法同时对5个浓度的PEDV cDNA模板进行检测,比较检测结果,评估本研究建立的ddPCR检测方法的敏感性。
1.6 ddPCR检测方法的特异性试验
采用本研究建立的ddPCR方法对PEDV、PDCoV、PRV、TGEV等12种全基因组核酸总RNA反转录cDNA同时进行检测,以评估该ddPCR检测方法的特异性。
1.7 ddPCR检测方法的重复性试验
选取浓度分别为1.0×103、1.0×102、1.0×101copies·μL-1PEDV核酸 cDNA为模板,用本研究建立的ddPCR方法进行扩增,对每个浓度进行3批次批间试验和批内试验检测,计算变异系数,评估该方法的重复性。
1.8 ddPCR检测方法对临床样品的检测
采用本研究建立的ddPCR方法和行业标准FQ-PCR方法对实验室保存150份临床疑似PEDV感染的肠内容物、小肠组织、粪便等样品进行检测,将1.0×104copies·μL-1PEDV核酸cDNA模板作为阳性对照,以正常vero细胞培养物作为阴性对照,评价ddPCR方法对临床样品检测效果。
2 结 果
2.1 ddPCR反应程序的优化
2.1.1 ddPCR预混酶的选择 根据两种预混酶推荐的引物对/探针浓度进行扩增,结果显示:采用2×qScriptTMXLT 1-Step RT-qPCR ToughMix®的拷贝数为7 338 copies·μL-1;采用2×PerfeCTa® qPCR ToughMix® UNG的拷贝数为10 647 copies·μL-1。因此,选择2×PerfeCTa® qPCR ToughMix® UNG作为本研究的预混酶。
2.1.2 ddPCR引物对/探针浓度以及退火温度的优化 ddPCR引物对/探针浓度的优化结果显示:上下游引物浓度各0.5 μmol·L-1,探针浓度0.25 μmol·L-1时拷贝数最高,检测结果最稳定。最佳反应体系:上下游引物浓度各0.5 μmol·L-1,探针浓度为0.25 μmol·L-1,2×PerfeCTa® qPCR ToughMix® UNG 12.5 μL,Fluorescein sodium Salt 2.5 μL,PEDV模板3 μL,无菌蒸馏水补足至总体积25 μL。ddPCR退火温度的优化结果显示:退火温度为56 ℃时,阴阳性微滴分区最为明显,拷贝数最高。最佳的反应程序:45 ℃ 5 min,95 ℃ 3 min;95 ℃ 15 s,56 ℃ 45 s 共45个循环。
2.2 ddPCR检测方法的评价
2.2.1 与行标FQ-PCR方法敏感性比较试验 行业标准FQ-PCR方法的标准曲线结果显示,相关系数R2为0.993,斜率为-3.647。本研究建立的ddPCR方法线性动态灵敏度范围和标准曲线:y=-1.045 3x+4.193,R2为99.8%(图1)。ddPCR方法检测极限为0.15 copies·μL-1(图2),行业标准FQ-PCR方法检测下限为10 copies·μL-1。结果表明,本研究建立的ddPCR方法敏感性明显高于行业标准FQ-PCR方法。
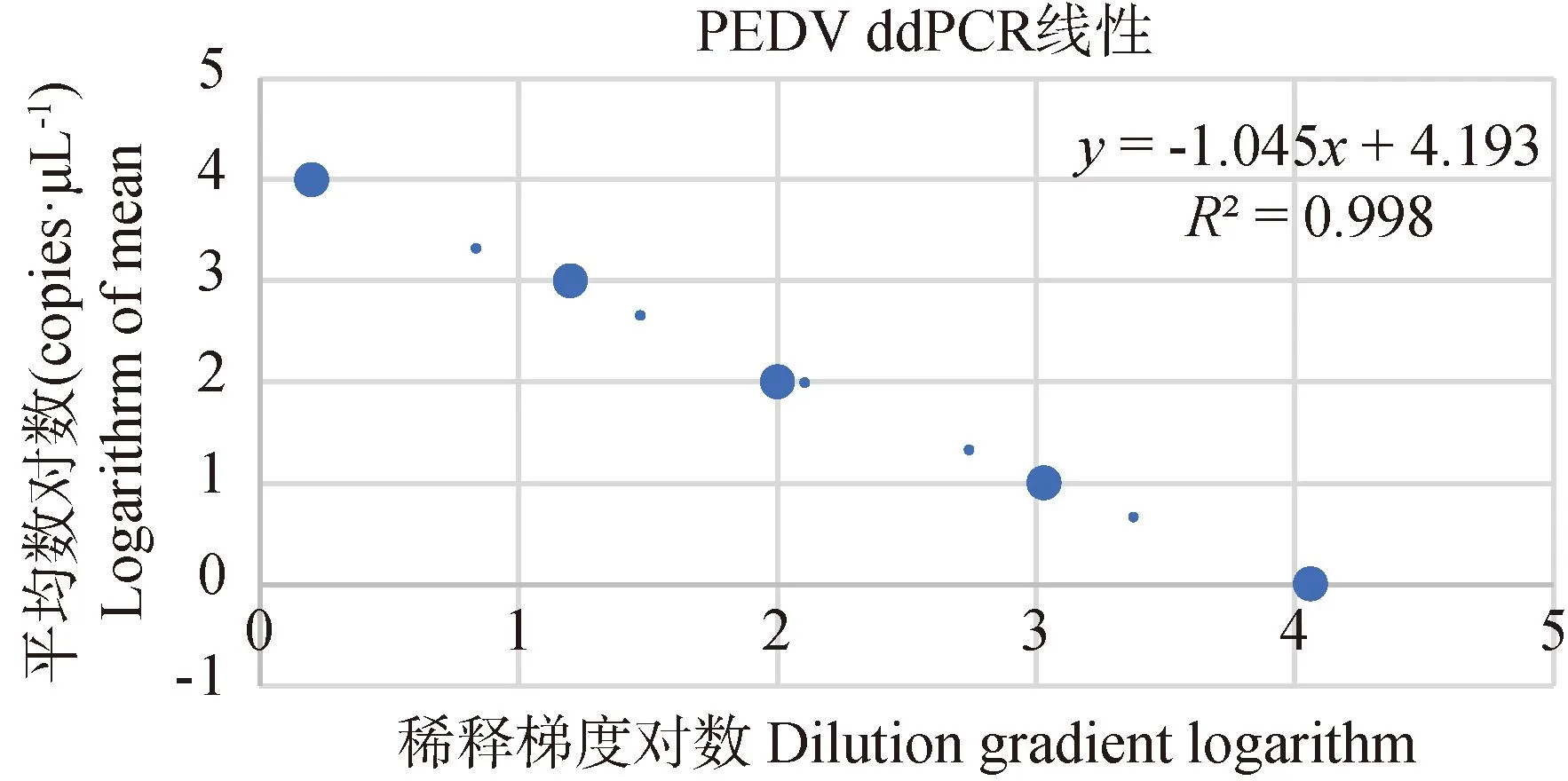
图1 ddPCR方法对PEDV检测的标准曲线Fig.1 Standard curve of PEDV detection by ddPCR method
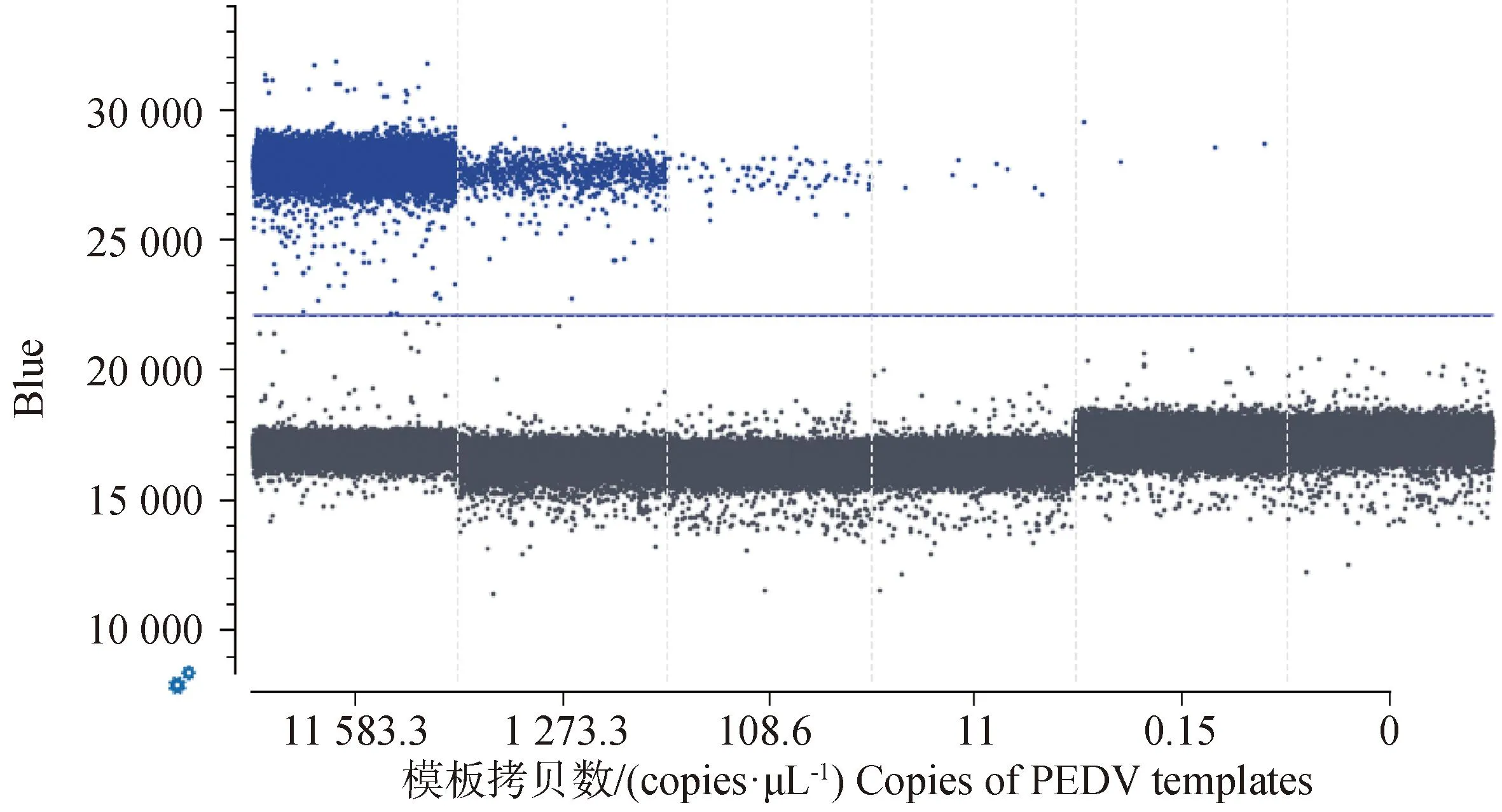
图2 ddPCR方法对10倍梯度稀释的PEDV检测Fig.2 Detection of PEDV with 10-fold gradient dilution by ddPCR
2.2.2 特异性试验 使用本研究建立方法,仅PEDV阳性对照出现扩增,拷贝数为10 087 copies·μL-1,阴性对照以及PDCoV、PRV、TGEV等11种全基因组核酸均未出现阳性微滴。结果表明,本研究建立的ddPCR方法特异性强。
2.2.3 重复性试验 结果显示,批内重复性试验变异系数(CV)为2.10%~7.40%;批间重复性试验变异系数(CV)为1.52%~7.40%(表4)。结果表明,本研究建立的ddPCR方法重复性好。
2.2.4 ddPCR检测方法对临床样品的检测 本研究建立的ddPCR方法和行业标准FQ-PCR方法对150份临床样品的检测结果显示:行业标准FQ-PCR方法检出76份阳性样品,检出率为50.6%;ddPCR方法检出89份阳性样品,检出率为59.3%(其中76份为行业标准FQ-PCR方法检测阳性样品,13份为行标FQ-PCR方法检测阴性样品);其中,ddPCR方法检出的63份阳性样品中的48份与标准荧光PCR检测阳性结果一致。结果表明,本研究建立的ddPCR方法灵敏性高,具有很好的临床应用效果,可以有效避免漏检。
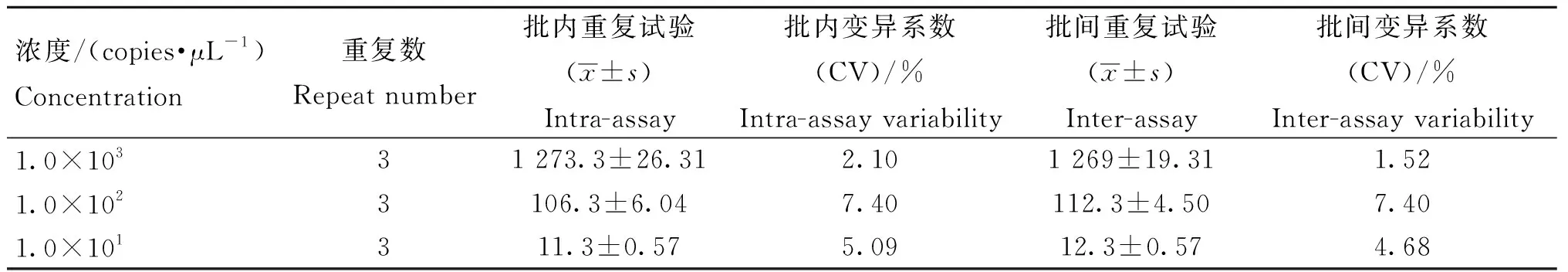
表3 ddPCR方法检测PEDV的重复性试验Table 3 Repeatability test of ddPCR method for PEDV detection
3 讨 论
近年来,非洲猪瘟、猪流行性腹泻、猪丁型冠状病毒病、猪伪狂犬病等猪重要病毒性疫病严重威胁着我国养猪业健康发展和公共卫生安全[11]。由PEDV变异毒株引起的猪群腹泻性疫病是当前除非洲猪瘟以外严重影响我国生猪养殖的重要疫病[12]。目前对PEDV感染进行快速、敏感的病原学检测的常用手段是FQ-PCR方法,其存在敏感性低、判读需依赖Ct值等问题。建立PEDV ddPCR方法对PEDV感染的早期诊断、PEDV核酸标准物质的研制以及PEDV疫苗抗原含量评价等具有重要意义。
综合考虑到变异毒株和经典毒株的保守区域,本研究在设计引物对/探针时选择能涵盖变异毒株与经典毒株的M基因[13],以建立适用性更广的PEDV ddPCR方法。在设计引物的同时,严格控制退火温度之间的差距,保证上下游引物Tm值的差距最小。对退火温度进行优化,当退火温度为56 ℃时,PEDV阴阳性微滴分区最为明显,可疑微滴最少,拷贝数最高。证明引物之间有着较好的特异性、灵敏性和稳定性。
本研究对自建FQ-PCR方法和行业标准FQ-PCR方法[10]进行比较。结果显示,自建的FQ-PCR方法在检测的敏感性、重复性方面优于行业标准FQ-PCR方法。为了使ddPCR方法的检测定量更加准确,本研究选取敏感性强、重复性好的自建FQ-PCR方法的引物对/探针来建立ddPCR方法。本研究建立的ddPCR方法,极大地提高了检测的灵敏性,其特异性较强,与PDCoV、PRV等11种病毒均无交叉反应。当PEDV处于潜伏期感染时,病毒的含量较低,使用普通PCR方法和荧光RT-PCR方法很难检测到。而本研究建立的ddPCR方法最低检测极限为0.15 copies·μL-1,为PEDV早期检测提供了重要的技术手段。另外,临床样品的检测结果显示,ddPCR与行业标准FQ-PCR方法符合率为100%,敏感性高于行业标准FQ-PCR,能够满足PEDV的临床样品检测。
采用ddPCR方法和FQ-PCR方法对150份临床样品进行检测,结果显示,ddPCR方法对行业标准FQ-PCR方法检测阳性样品的检测符合率为100%。在PEDV感染早期及病毒含量较低的临床样品检测中,ddPCR方法更具有检测优势。
4 结 论
本研究基于PEDVM基因保守区域为模板设计引物对和探针,经优化反应体系及反应条件,建立了一种敏感性高、特异性强、重复性好、临床应用效果佳的PEDV ddPCR方法,为临床上PEDV感染的早期诊断和定量检测、PEDV核酸标准物质的研制以及猪流行性腹泻防控等提供了技术手段。



