胡 越,林传明
(1.赣南医学院2019级硕士研究生;2.赣南医学院第一附属医院血液科,江西 赣州 341000)
组织细胞坏死性淋巴结炎(Histiocytic necrotizing lymphadenitis,HNL),也称为菊池-藤本病(Kikuchi-fujimoto disease),最早由菊池[1]、藤本[2]两位日本学者报道,是一种罕见的淋巴组织细胞疾病。噬血细胞综合征(Hemophagocytic syndrome,HPS)分为原发性和继发性两种,继发性噬血细胞综合征(Hemophagocytic lymphohistiocytosis,HLH)不是一种独立的疾病,而是继发于多种致病因素导致单核巨噬细胞系统反应性增生,伴有吞噬自身血细胞的现象。由于此病发生率不高,易误诊为其他良性淋巴结病、淋巴瘤等。现将我科收治的1例组织坏死性淋巴结炎合并继发性噬血细胞综合征病例报道如下。
1 临床资料
患者女,19 岁,学生,因“发热9 天”于2021年3月14日入住我院。患者于12 天前开始出现乏力,9天前开始出现发热,以夜间为甚,最高39.3 ℃,伴畏寒、头晕、纳差、乏力,无盗汗、咳嗽、腹胀、腹痛、腹泻、肌肉酸痛等。自服“999感冒灵”后症状无明显好转,到当地诊所就诊,查血常规:白细胞2.9×109·L-1,血红蛋白115 g·L-1,血小板134×109·L-1,于2021年3月9日至于都县人民医院就诊,予帕拉米韦抗病毒,左氧氟沙星、多西环素抗感染等治疗后患者仍有反复发热,夜间明显,最高超过40 ℃,高热时感头晕,伴恶心、呕吐、纳差,偶有黑蒙。发病后患者精神状态、饮食、睡眠差。有地中海贫血病史和家族史,对“氨基比林”“克林霉素”“林可霉素”“青霉素”“头孢”过敏,余既往史、传染病接触史无特殊发现。入院查体:体温37.4 ℃,脉搏117次·min-1,呼吸20次·min-1,血压107/69 mmHg。颈部可触及多枚肿大淋巴结,质软,活动度可,伴或不伴触痛,最大者位于左侧颈后,约1.0 cm×2.0 cm,余浅表淋巴结未触及,扁桃体Ⅱ度肿大,心肺查体无异常,腹软、无压痛,肝脾未及,双下肢无水肿。血常规:白细胞0.73×109·L-1,血红蛋白94 g·L-1,血小板102×109·L-1,中性粒细胞0.29×109·L-1,淋巴细胞0.36×109·L-1。血生化:甘油三酯0.96 mmol·L-1,纤维蛋白原2.61 g·L-1,空腹血糖5.59 mmol·L-1,铁蛋白411 ng·mL-1,乳酸脱氢酶(LDH)600 U·L-1,α-羟丁酸脱氢酶472 U·L-1;C 反应蛋白(CRP)9.09 mg·L-1;白介素1 025.47 pg·mL-1,干扰素γ 249.56 pg·mL-1,白介素2、4、6、肿瘤坏死因子正常;风湿免疫相关检查阴性、结核抗体阴性、血培养阴性。淋巴结彩超:双侧颈部可见数个低回声淋巴结,右侧大者17 mm×3.1 mm,左侧大者22 mm×19.9 mm;左侧锁骨下见16 mm×4 mm 低回声淋巴结,双侧锁骨上见数个低回声淋巴结,左侧大者18.9 mm×7 mm,右侧大者12 mm×4 mm。脾脏:长径115 mm,厚径42 mm,肝(-);胸腹部CT 检查未见明显异常。3月14日患者入院后,我们结合患者反复发热的病史,且伴有粒细胞缺乏,已使用多种抗生素治疗效果欠佳,依据《中国中性粒细胞缺乏伴发热患者抗菌药物临床应用指南(2020年版)》[3],予以亚胺培南西司他汀抗感染、补液、纠正电解质紊乱等治疗。治疗期间,患者3月17日出现腹痛,具体位于脐周左上方及右下方,压痛明显,无反跳痛,排便后缓解;3月19日夜间开始出现全身红疹,伴瘙痒,数小时后可自行消退,但反复发作且逐渐连接成片状,考虑过敏引起,予以炉甘石洗剂治疗;治疗期间患者体温峰值逐日下降,2021年3月19日开始体温不超过37.3 ℃,3月20日之后体温峰值不超过37.0 ℃(仅住院第1 天和第2 天使用了非甾体类抗炎药对症退热,之后未使用非甾体类抗炎退热药物)。但在单独使用抗生素治疗期间,患者乏力、纳差症状并无明显改善。在此治疗期间骨髓细胞学检查:(1)网状细胞偶伴吞噬现象(图1);(2)巨核细胞轻度成熟障碍;(3)外周血三系减少。NK 细胞活性(%):11.48(参考值≥15.11),△CD107a:1.78%(△CD107a≤5%:脱颗粒功能缺陷),△MFI:1.8(△MFI≥2.8 提示脱颗粒功能正常),可溶性白细胞介素2 受体(sCD25):1 294.0 U·mL-1。2021年3月19日赣南医学院第一附属医院淋巴结活检:(颈部淋巴结)送检淋巴结镜下见淋巴结结构大致保存,可见多灶斑驳淡染区,淡染区内可见坏死、核碎屑和组织细胞,副皮质区呈反应性增生改变,结合形态及免疫组化符合组织细胞坏死淋巴结炎(图2)。淋巴结活检免疫组化:CD3T 细胞(+)、CD5T 细胞(+)、CD7T 细胞(+)、CD8T 细胞(+)、CD68 组织细胞(+)、CD123(-)、S-100(-)、Ki-67 约30%(+)、CD20B 细胞(+)、ALK(-)、CD30(-)、CD21 示(FDC+)、CD23示(FDC+)、MPO(+)、CD2T 细胞(+)、CD4T 细胞及组织细胞(+)、EBER(-)、PAX-5B 细胞(+)。2021年3月22日临床确诊为组织细胞坏死性淋巴结炎合并噬血细胞综合征,并开始在原有治疗方案基础上,加用糖皮质激素(40 mg甲泼尼龙2次·d-1)静滴。3 天后患者乏力、纳差、腹痛、红疹、恶心、呕吐的症状均逐渐好转。复查血常规:白细胞7.42×109·L-1,红细胞4.35×1012·L-1,血红蛋白98 g·L-1,血小板329×109·L-1,中性粒细胞4.64×109·L-1,淋巴细胞2.16×109·L-1,网织红细胞87.9×109·L-1,网织红细胞比例2.02%;乳酸脱氢酶233 U·L-1,α-羟丁酸脱氢酶175 U·L-1,铁蛋白63.69 ng·mL-1;白介素10 0.7 pg·mL-1,干扰素γ 0.98 pg·mL-1。对比入院时白细胞明显升高,血红蛋白较前降低,乳酸脱氢酶、α-羟丁酸脱氢酶、铁蛋白、C 反应蛋白、细胞因子较前明显降低,查体提示肿大的淋巴结较前缩小,压痛消失。患者要求出院,门诊继续服用泼尼松片20 mg·d-1,服用1 周后每天减少5 mg 直至减停,1月后随访患者完全康复。
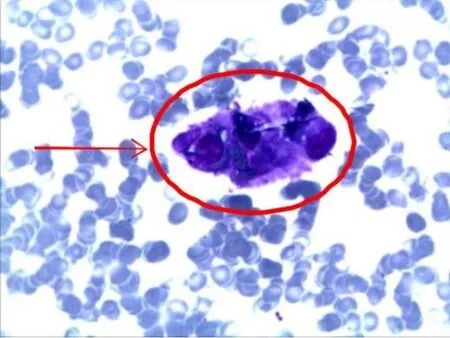
图1 骨髓细胞学图(瑞氏染色×100)
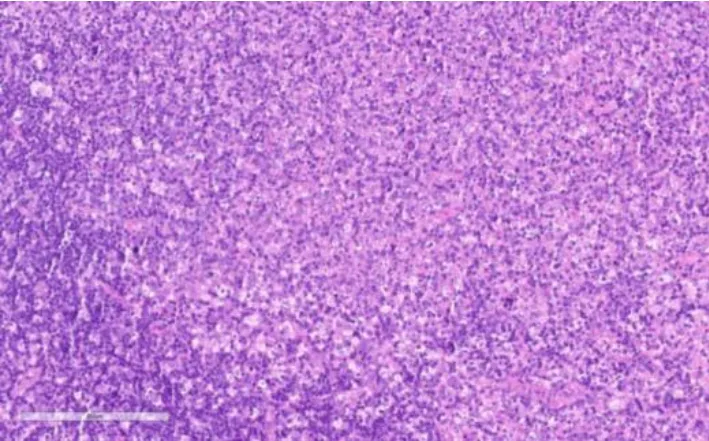
图2 淋巴结活检病理图(HE×40)
患者随访至今,生活与正常人无异,未出现发热、纳差、全身乏力、腹痛等症状,病情无复发倾向。但自诉偶有出现皮肤瘙痒,伴有皮疹,服用氯雷他定后可缓解。
2 讨 论
HNL 病程表现为良性自限性疾病,疾病病程通常在1~4 个月,以亚裔青年女性多见。临床表现主要包括抗生素治疗无效的发热、乏力、淋巴结肿大等,淋巴结肿大又以颈后三角区多见[4]。HNL 的实验室检查常表现为白细胞减少、中性粒细胞减少,ESR、CRP、LDH、铁蛋白升高[5-6]。这些均缺乏特异性,且因此病发病率不高[4],故在临床实践中难以优先考虑,常在淋巴结活检后发现。病因和发病机制方面,主流观点认为病因是感染和自身免疫紊乱。HNL 的临床表现类似于病毒感染,对抗生素不敏感,且组织病理学特征(即皮质旁扩张、免疫母细胞增殖、副皮质坏死、T 细胞占优势以及外周血中不典型/反应性淋巴细胞)与病毒感染相似,故而认为病因与病毒感染相关[7-9]。HNL也被认为与一些自身免疫性疾病有关,尤其是系统性红斑狼疮(SLE)[9-10]。HNL 刺激淋巴细胞和组织细胞胞浆与已知的系统性红斑狼疮和其他自身免疫性疾病患者的内皮细胞和淋巴细胞的结构相似。有研究表明,30%患者在系统性红斑狼疮发作之前就被诊断为HNL,47%患者HNL 和SLE 同时发生或诊断,23%的患者HNL的诊断之前已经有SLE的病史[11]。
噬血细胞综合征,又称噬血细胞性淋巴组织增多症,是一种复杂的、威胁生命的全身性过度炎症综合征,其特征是持续发热、细胞减少、肝脾肿大、凝血障碍和典型生物标志物的升高,包括铁蛋白和可溶性白细胞介素2 受体(sIL-2R)。患者还可能出现皮疹、肝炎、弥散性血管内凝血、急性肝功能衰竭、中枢神经系统(CNS)受累、多器官衰竭和其他问题,包括死亡。目前诊断噬血细胞综合征通用的标准,称为HLH-2004诊断标准[12-13]。
HNL 的总体预后良好,大多数病例只需要支持性治疗,在几周到几个月内自然完全康复,故有学者认为此病不需要任何治疗;但对于病情严重尤其是合并有其他疾病的情况,如合并有DIC[14]、噬血细胞综合征[15]、继发于血小板减少所致的脑出血等,则有可能危及生命[4],需药物干预疾病进程。另外,初诊的HNL 患者也有合并系统性红斑狼疮[16]、周围神经病变[17]、血管炎[18]等自身免疫相关性疾病,也被普遍认为需积极治疗。根据已有文献报道,此病对糖皮质激素敏感度高、疗效确切,是最常用的药物[17]。此外,羟氯喹、免疫球蛋白在HNL 的治疗方面也取得了令人满意的效果[17-18]。在远期的随访中发现,此病可能复发或进展为SLE[10]。
由于感染是发热最常见的病因,故本例患者在诊断尚不完全明确时就已经且继续被予以多种抗菌药物治疗;之后,骨髓血涂片及骨髓活组织检查发现吞噬细胞现象,此为噬血细胞综合征的诊断标准之一[12]。继续完善NK 细胞活性及sCD25 检查,结合凝血分析、铁蛋白、血常规、肝功能、腹部彩超检查及病史,依据HLH-2004 诊断标准诊断为噬血细胞综合征。HNL 疾病本身就符合HLH 的部分诊断标准,如发热、脾大、血细胞减少、铁蛋白升高[6],NK细胞活性及sCD25由于并非常用的检查指标,故难以判断HNL 疾病本身是否会发生上述实验室结果异常。作为两种与淋巴系统密切相关的疾病,有学者认为,它们可能构成疾病连续性的一部分,而不是独立的个体[12]。HNL合并HLH 并非罕见现象,在一次样本量为51 例的回顾性研究中,有3 例患者并发有噬血细胞综合征,而这51 例样本中只有22例完善了骨髓细胞学检查[19]。本例患者有8项标准中的5 项:发热、脾大、骨髓涂片和活组织中存在吞噬血细胞现象、血细胞二系减少、NK 细胞活性减低。铁蛋白呈进行性升高、甘油三酯出现一过性升高但均不足以达到标准,凝血功能一直处于正常范围。此后在血常规和血生化指标的部分指标持续检测过程中,发现患者LDH、铁蛋白、CRP 呈进行性升高趋势,在使用激素前达到峰值,使用激素治疗后明显降低,且在使用激素2周内均回到正常水平,这与患者的临床症状相一致。笔者推测,这些异常指标虽然缺少特异性,但由于创伤小,可多次进行定量检测,有望作为反应病情变化的指标。
由于HNL 病在临床上并不常见,但在临床上发现持续发热的患者,尤其是抗生素不能缓解的发热者,应当:(1)进行全面的体格检查,尤其包括浅表淋巴结的检查;(2)对于存在淋巴结肿大者必要时可积极完善淋巴结活检以确诊或排除HNL、淋巴瘤、结核性淋巴结炎等疾病,以避免患者承受过多的药物不良反应和疾病本身的痛苦;(3)HNL 可并发HLH、SLE 等,故在确诊后需积极完善骨髓细胞学、dsDNA 等检查以制定对应的治疗方案。此外,目前对此病的了解尚不完全,对其病因、诊断及治疗均无统一标准,仍需后续的研究及总结。



