胡红美,郭远明,*,雷 科,张小军,严忠雍,何依娜,尤炬炬,刘 琴
(1.浙江省海洋水产研究所 浙江省海水增养殖重点实验室,浙江 舟山 316100;2.浙江海洋学院水产学院,浙江 舟山 316000)
分散固相萃取净化-气相色谱法测定水产品中氯霉素和氟苯尼考
胡红美1,郭远明1,*,雷 科2,张小军1,严忠雍1,何依娜1,尤炬炬1,刘 琴1
(1.浙江省海洋水产研究所 浙江省海水增养殖重点实验室,浙江 舟山 316100;2.浙江海洋学院水产学院,浙江 舟山 316000)
建立测定水产品中氯霉素和氟苯尼考的气相色谱-电子捕获检测方法。样品通过乙酸乙酯萃取、正己烷初步净化后,经过分散固相萃取进一步净化、硅烷化试剂衍生,再采用气相色谱-电子捕获法检测、外标法定量。结果表明:氯霉素、氟苯尼考分别在1.5~100 μg/L、6~400 μg/L范围内,组分含量与峰面积呈线性相关,相关系数分别为0.998 1和0.999 7,检出限分别为0.1、0.3 μg/kg。氯霉素和氟苯尼考在不同基质的水产品(鲫鱼、青蟹、南美白对虾)中不同加标水平回收率分别为82%~106%、87%~111%和91%~98%,相对标准偏差分别为2.3%~4.9%、2.4%~4.5%和1.4%~4.1%(n=5)。本方法基体干扰小、线性范围宽,灵敏度、准确度、精密度能满足水产品中氯霉素类药物的含量分析。
分散固相萃取;气相色谱-电子捕获检测法;水产品;氯霉素类药物
氯霉素、氟苯尼考同属于氯霉素类药物,是一种广谱抗菌药,因其具有抑菌性和杀菌性被广泛用于水产养殖过程中细菌性疾病的预防和治疗。氯霉素因其苯环上有硝基,故对人体有严重的副作用,会抑制骨髓造血功能、引起再生障碍性贫血[1]。欧盟、美国以及我国等国规定在食品中氯霉素零容许量,即不得检出。氟苯尼考在安全性和有效性方面虽比氯霉素具有明显优势,但由于兽医临床的滥用、误用,细菌耐药性问题越来越严重,我国规定鱼肌肉+皮部分最高残留限量为1 000 μg/kg,欧盟也规定鳍鱼类肌肉+皮部分最高残留限量为1 000 μg/kg。
目前,国内外关于氯霉素类药物残留检测方法已很很多,主要采用酶联免疫法[2-5]、气相色谱法[6-10]、气相色谱-质谱法[11-13]、液相色谱法[14-16]、液相色谱-串联质谱法等[17-25]。酶联免疫法使用酶联免疫检验试剂盒,通常适合于大规模筛选,但由于抗体批次不同,测定结果将出现一定差异、重复性差,而且特异性很强,不能进行多残留检测,试剂昂贵,易出现假阳性结果[26-27];气相色谱法、气相色谱-质谱法一般需要衍生试剂进行衍生化[28],方法较繁琐;液相色谱法灵敏度达不到要求[29];高效液相色谱-串联质谱法背景干扰小、灵敏度高,是氯霉素类药物残留检测的首选方法[30],也是目前氯霉素类药物检测方法的研究热点,但液相色谱-串联质谱仪价格昂贵,难以一般实验室普及。而采用气相色谱法测定氯霉素类药物近2年几乎未见报道,且以前报道中均采用固相萃取法进行再净化,使得操作更加复杂。因此研究一种更简单的前处理方法对改善气相色谱法的应用前景显得十分重要。
随着分散固相萃取近年来在药物残留前处理中发挥的巨大作用[31-33],目前已有学者结合快速、简易、廉价、有效、稳定、安全(quick, easy, cheap, effective, rugged, safe,QuEChERS)前处理法、采用液相色谱-串联质谱法测定氯霉素[34]。但这些报道首先限定了分散溶剂,再选择合适的固相吸附剂,忽略了分散溶剂对不同吸附剂的影响,此外有些固相吸附剂在不同分散溶剂中的效果截然相反。本研究通过对分散固相萃取方法的研究,首次建立了分散固相萃取-气相色谱法测定水产品中氯霉素和氟苯尼考残留量的方法。
1 材料与方法
1.1 材料与试剂
所有水产品(鲈鱼、大黄鱼、草鱼、鲫鱼、鲤鱼、鳊鱼、甲鱼、青蟹、南美白对虾)采集于舟山、温州和台州等地的无公害水产养殖基地。
正己烷、丙酮、乙酸乙酯、甲醇(均为色谱纯)美国Merck公司;氯霉素、氟苯尼考(纯度均>98%)德国Dr. Ehrenstorfer公司;N,O-双(三甲基硅烷基)三氟乙酰胺∶三甲基氯硅烷(99∶1)(衍生化试剂) 美国Regis公司;其他试剂均为分析纯。
1.2 仪器与设备
GC-450气相色谱仪(配有电子捕获检测器) 美国Varian公司;Centrifuge 5810高速离心机 德国Eppendorf公司;R-215旋转真空蒸发仪 瑞士Büchi公司;KD200可视氮吹仪(配有5 mL模块) 杭州奥盛仪器有限公司;N-丙基乙二胺吸附剂(primary secondary amine,PSA,粒径50 μm)、C18吸附剂(粒径50 μm)上海安谱科学仪器有限公司。
1.3 方法
1.3.1 样品前处理
1.3.1.1 制样
鱼:去鳞、去皮,沿脊背取肌肉;蟹、甲鱼:取可食肌肉部分;虾:去头、去壳、去附肢,取可食肌肉部分。样品切为不大于0.5 cm×0.5 cm×0.5 cm的小块,用高速组织捣碎机匀质,密封,-18℃冷冻保存。
1.3.1.2 提取
将样品解冻,称取5.00 g于50 mL离心管中,加入20 mL乙酸乙酯,涡旋2 min,以6 000 r/min高速离心3 min,收集提取液于100 mL梨形瓶中,再往试样中加入10 mL乙酸乙酯,重复上述过程1次,合并提取液,于40 ℃水浴中减压旋转蒸发至干。
1.3.1.3 净化
残渣用1 mL甲醇溶解后,加入25 mL 4%氯化钠溶液,涡旋30 s,再加入15 mL正己烷,涡旋2 min,以6 000 r/min高速离心3 min,弃去上层正己烷相,再用10 mL正己烷重复提取1遍(如果试样含脂肪较多,可重复提取多遍)。水相中加入15 mL乙酸乙酯,涡旋2 min,以6 000 r/min高速离心3 min,吸取乙酸乙酯相,过无水硫酸钠脱水过滤于50 mL梨形瓶中,再向水相中加入10 mL乙酸乙酯,重复上述操作1次。用少量乙酸乙酯淋洗无水硫酸钠,合并提取液,于40 ℃水浴中减压旋转蒸发至干。加入2 mL丙酮溶解提取物并转移到5 mL具塞玻璃离心管中,用1 mL丙酮洗涤梨形瓶,合并丙酮溶解液,加入100 mg PSA吸附剂,涡旋30 s,以3 000 r/min高速离心3 min,吸取上清液于另一洁净5 mL具塞玻璃离心管中,于50 ℃干浴中吹氮蒸发至近干,再用1 mL丙酮洗涤离心管壁并吹氮蒸发至干。
1.3.1.4 衍生
再加入100 μL衍生化试剂,涡旋混合30 s,在70℃烘箱中反应30 min后,再于50 ℃干浴中吹氮蒸发恰好至干。加入100 μL水,再加入1 mL甲苯,涡旋混匀,静置10 min,以4 000 r/min高速离心3 min,吸取上层甲苯相,取1 μL进入气相色谱分析。
1.3.2 色谱条件
色谱柱:D B-5 M S毛细管气相色谱柱(30 m×0.25 mm,0.25 μm);不分流进样;载气:高纯氮气(99.999%);流速2.0 mL/min;进样口温度260 ℃;电子捕获检测器温度300 ℃;升温条件:柱初始温150 ℃,保持1.0 min,以15 ℃/min升至260 ℃,再以30℃/min升至280 ℃,保持5.0 min;总运行时间24 min;进样量1 μL。
1.3.3 标准溶液配制
分别准确称取氯霉素和氟苯尼考标准品0.005 0 g,用甲醇溶解并定容至10 mL容量瓶中,配制成500 mg/L标准储备液。再通过逐级稀释、甲醇定容,配制成氯霉素质量浓度为100 μg/L、氟苯尼考质量浓度为400 μg/L的混合标准使用液,4 ℃储存。
1.3.4 标准曲线的绘制
分别量取15、25、50、100、200、500、1 000 μL的混合标准使用液,于50 ℃干浴中吹氮蒸发至干,加入100 μL衍生化试剂,涡旋混合30 s,在70 ℃烘箱中反应30 min后,再于50 ℃干浴中吹氮蒸发恰好至干。加入1 mL甲苯,再加入100 μL水,涡旋混匀,静置10 min,以4 000 r/min高速离心3 min,吸取上层甲苯相,配制成质量浓度为1.5、2.5、5、10、20、50 μg/L和100 μg/L的氯霉素溶液、质量浓度为6、10、20、40、80、200、400 μg/L的氟苯尼考标准工作液,供分析。以峰面积为纵坐标、氯霉素和氟苯尼考质量浓度为横坐标,绘制标准曲线。
1.4 样品定量分析
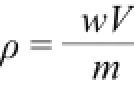
式中:ρ为测定含量/(μg/kg);w为上机质量浓度/(μg/L);V为样品定容体积/mL;m为取样质量/g。其中V=1 mL,m=5.00 g。
2 结果与分析
2.1 提取溶剂的选择
氯霉素、氟苯尼考微溶于水,易溶于乙酸乙酯、甲醇、乙腈等有机溶剂。但甲醇、乙腈对人体毒性较大,且乙腈在挥干时容易冒泡,挥干时间长[12],通常用于奶制品的提取;甲醇的极性比较大,提取液含杂质较多;而乙酸乙酯偏中性,提取液含杂质较少,提取效率高,毒性小[8],容易挥干。本实验采用乙酸乙酯作为提取溶剂可以最大限度的从水产品中提取出有效成分并使共萃取的干扰物较少。
2.2 净化条件的确定
对于某些样品,经正己烷去除脂质和类脂后,无需进一步净化便可满足分析方法要求,但对于净化不完全的样品则需要进行进一步净化。文献报道比较多的是采用C18固相萃取柱[6-7,19]、硅胶固相萃取柱[14]、HLB固相萃取柱[25],或者多种固相萃取柱相结合[13]的方法。虽然固相萃取法能达到较好的净化效果,但需要进行萃取柱活化、上样、淋洗、洗脱等过程,步骤较多、过柱时间较长,过柱和洗脱过程中流速控制不当容易造成样品损失,且成本较高,不利于大批量样品的检测。本实验采用分散固相萃取净化,比较了100 mg PSA吸附剂、100 mg C18吸附剂在不同有机溶剂(乙酸乙酯、甲醇、丙酮)中的净化效果(表1),结果表明,C18吸附剂在不同有机溶剂(乙酸乙酯、甲醇、丙酮)中的净化效果均不明显;PSA吸附剂在乙酸乙酯中可完全吸附目标化合物,使得最终检测结果中无氯霉素和氟苯尼考,此外PSA吸附剂在甲醇和丙酮中净化效果均较好。这可能是因为PSA吸附剂能有效地去除食品中影响农残检测的碳水化合物、脂肪酸、有机酸、酚类、一些极性色素以及糖类等干扰,而C18吸附剂主要用于去除脂肪和酯类等非极性干扰物,在前一步正己烷净化中已除去大部分的脂质和类脂。考虑到甲醇毒性较大且沸点较高,选择用丙酮溶解提取物,在丙酮中用PSA吸附剂净化。此外,考察了不同量(0~200 mg)的PSA吸附剂对净化效果的影响(表1)。结果表明,随着PSA吸附剂量的增加,净化效果有所增加,但由于PSA吸附剂会吸附一定的丙酮,PSA剂量越高,回收率会有所下降,最终选择使用100 mg PSA吸附剂,此时既能达到较好的净化效果,又能保证较高的回收率。
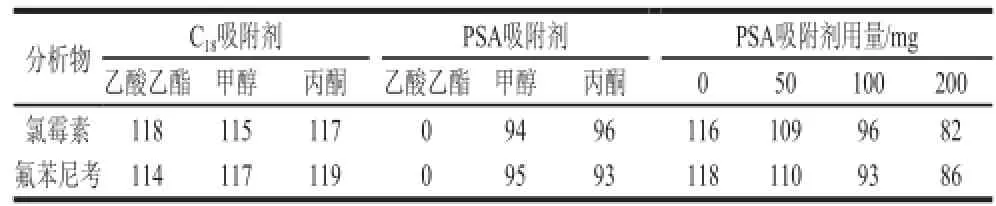
表1 不同剂量的C18、PSA吸附剂在乙酸乙酯、甲醇、丙酮中的净化回收率(n=5)Table 1 Recoveries of CAP and FF purified with C18vs. PSA in ethyl acetate, methanol or acetone as well as different amounts of PSA in acetone (n = 5)%
2.3 衍生条件的确定
由于氯霉素、氟苯尼考含有羟基、氨基、硫酰基及亚胺基,都有很强的极性且热稳定性差、难挥发,在采用气相色谱法分析此类化合物时,需对其极性官能团进行酯化、硅烷化或酰化,生成热稳定性和易挥发的衍生物。本实验利用硅烷化试剂将氯霉素、氟苯尼考的羟基硅烷化衍生成易挥发、热稳定的物质。由于衍生化试剂遇水易分解,会造成目标化合物衍生失败,且实验证实若衍生时存在少量水时,将检测不到目标物。因此必须保证衍生过程中无水。此外,衍生完毕后,多余的衍生溶液必须通过吹氮蒸发恰好至干,过头或未干均会影响衍生效果,降低重复性。因此,本实验选用可视氮吹仪随时观察氮吹过程。此外,本实验在衍生化后加100 μL水灭活,以终止在进样时衍生化试剂与分析物在进样口进一步发生反应,保证了测定结果的稳定性,同时也净化了反应液,有利于色谱峰的分离。实验结果表明,如果不加100 μL水灭活,直接加有机试剂(如正己烷)定容,对氯霉素影响不大,峰面积变化不超过10%,但对氟苯尼考影响甚大,会造成氟苯尼考色谱峰面积不稳定(随着时间延长,先变大,后变小),峰面积变化甚至会超过50%。
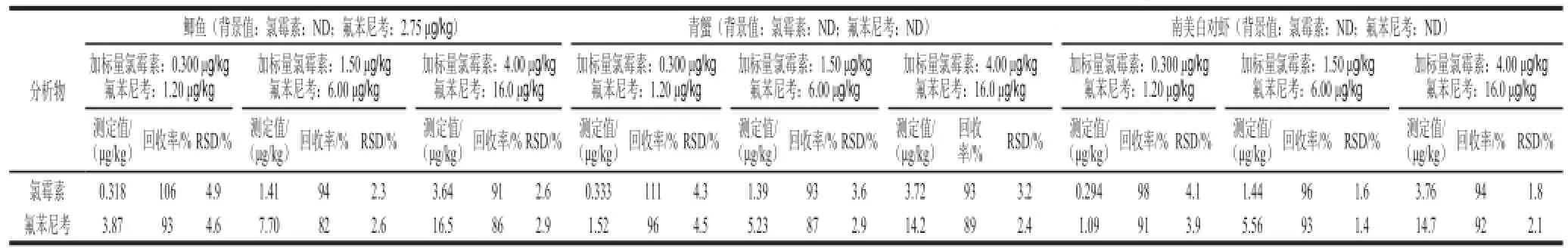
表3 鲫鱼、青蟹、南美白对虾中氯霉素和氟苯尼考的加标回收率及方法的精密度(n=5)Table 3 Recoveries and RSDs of CAP and FF in spiked crucian, blue crab, and Penaeus vannawei by the proposed method (n = 5)
2.4 定容试剂的选择
本实验比较了正己烷、甲苯定容对测定结果的影响。结果表明,同等条件下,采用甲苯定容可使氯霉素、氟苯尼考的色谱响应值增大,其中氯霉素峰面积增加5%~10%左右,而氟苯尼考增加将近40%~50%。高沸点的甲苯可提高实验重复性,放置一段时间后重新进样时的测定结果比正己烷稳定。因此,本实验采用衍生化结束后直接加100 μL水、1 mL甲苯涡旋离心后取甲苯相进样的定容方式。
2.5 方法评价
2.5.1 方法的线性范围及检出限
将标准系列溶液,按照1.3.2节进行分析。用外标法绘制出校正曲线。氯霉素、氟苯尼考分别在1.5~100、6~400 μg/L范围内与其峰面积呈良好线性关系,氯霉素、氟苯尼考检出限分别为0.1、0.3 μg/kg(表2)。

表2 回归方程、线性范围、相关系数和检出限Table 2 Regression equations, linear ranges and detection limits

图1 氯霉素(20 μg/L)和氟苯尼考(80 μg/L)标准品色谱图Fig.1 Standard chromatogram of 20 μg/L CAP and 80 μg/L FF
2.5.2 方法的精密度和回收率
为考察基体效应以及验证方法准确性和重复性,分别准确称取鲫鱼、青蟹、南美白对虾样品5.00 g,按照前述处理方法平行测定3 次,均未检出氯霉素,但在鲫鱼中检测出氟苯尼考,含量为2.75 μg/kg,相对标准偏差(relative standard deviation,RSD)为3.7%。再分别往5.00 g鲫鱼、青蟹、南美白对虾样品中各加入15、75 μL和200 μL的混合标准使用液,配制成低(氯霉素0.3 μg/kg、氟苯尼考1.2 μg/kg)、中(氯霉素1.5 μg/kg、氟苯尼考6 μg/kg)和高(氯霉素4 μg/kg、氟苯尼考16 μg/kg)3个质量浓度的加标样品,平行测定5 次。鲫鱼、青蟹、南美白对虾样品加标回收率为分别为82%~106%、87%~111%和91%~98%,RSD分别为2.3%~4.9%、2.4%~4.5%和1.4%~4.1%(n=5)。结果表明,该方法基体效应可以忽略,精确度和准确度满足分析方法要求。按照本分析方法,对采集的其他样品进行了测定,除了在草鱼中检测出氟苯尼考(含量为0.786 μg/kg),其他样品中均未检出氯霉素和氟苯尼考。
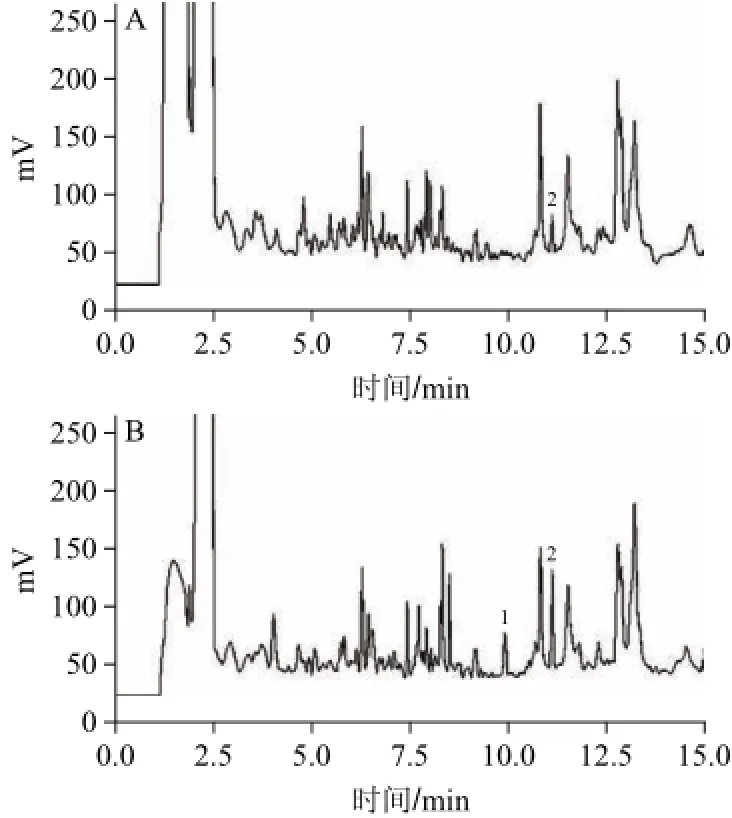
图2 鲫鱼样品(A)和加标鲫鱼样品(氯霉素1.5 μg/kg、氟苯尼考6.0 μg/kg)(B)气相色谱图Fig.2 Chromatograms of (A) crucian and (B) spiked crucian with 1.5 μg/kg CAP and 6.0 μg/kg FF
3 结 论
本实验采用分散固相萃取净化和气相色谱电子捕获法对水产品中氯霉素和氟苯尼考进行了同时测定。在进行分散固相萃取净化时,详细考察了不同固相吸附剂在不同分散溶剂中的净化效果,结果表明不同分散溶剂对固相吸附的影响很大。此外固相吸附剂的用量也会影响净化效果。最终选择100 mg PSA吸附剂、丙酮为分散溶剂。本研究还对衍生过程、定容试剂进行了优化,结果表明衍生过程必须无水,衍生完成后,多余衍生溶液必须恰好氮吹至干,并使用100 μL水灭活,再加入定容试剂甲苯。该方法灵敏、重复性好、准确度高、回收率令人满意,基体干扰小,能满足水产品中氯霉素类药物的分析要求,可进一步应用到其他食品中氯霉素类药物的检测。
[1] TURTON J A, HAVARD A C, ROBINSON S, et al. An assessment of chloramphenicol and thiamphenicol in the induction of aplastic anaemia in the BALB/c mouse[J]. Food and Chemical Toxicology, 2000, 38(10): 925-938.
[2] 郝凯, 过世东, 胥传来. 水产品中甲砜霉素残留的CELISA检测方法研究[J]. 食品工业科技, 2006, 27(2): 186-189.
[3] 周宏琛, 朱涛, 闫秋成, 等. 竞争酶联免疫吸附方法检测氯霉素假阳性原因和质量控制的探讨[J]. 水产科学, 2007, 26(2): 123-124.
[4] 彭运平, 齐维, 唐海波, 等. 应用酶联免疫法检测鱼肉、蜂蜜中氯霉素的残留量[J]. 现代食品科技, 2010, 26(12): 1414-1417.
[5] 谭慧, 麦琦. 酶联免疫分析法测定水产品中氯霉素残留量[J]. 中国卫生检验杂志, 2010, 20(7): 1649-1650.
[6] PFENNINGA P, MADSON M R, ROYBAL J E, et al. Simultaneous determination of chloramphenicol, florfenicol, and thiamphenicol residues in milk by gas chromatography with electron capture detection[J]. Journal of AOAC International, 1998, 81(4): 714-720.
[7] PFENNING A P, ROYBAL J E, RUPP H S, et al. Simultaneous determination residues of chloramphenicol, florfenicol, florfenicolamine, and thiamphenicol in shrimp tissue by gas chromatography with electron capture detection[J]. Journal of AOAC International, 2000, 83(1): 26-30.
[8] 王建华, 陈世山. 同时测定鱼肉中氯霉素和甲砜霉素残留量的毛细管气相色谱法[J]. 分析测试学报, 2001, 20(3): 89- 91.
[9] DING S Y, SHEN J Z, ZHANG S X, et al. Determination of chloramphenicol residue in fish and shrimp tissues by gas chromatography with a microcell electron capture detector[J]. Journal of AOAC International, 2005, 88(1): 57-60.
[10] 吴明媛, 杨姝丽, 黎小正, 等. 毛细管电子捕获气相色谱法同时测定水产品中氯霉素、甲砜霉素和氟甲砜霉素残留[J]. 广西农业科学, 2009, 40(5): 567-570.
[11] NAGATA T, OKA H. Detection of residual chloramphenicol, florfenicol, and thiamphenicol in yellowtail fish muscles by capillary gas chromatography-mass spectrometry[J]. Journal of Agricultural and Food Chemistry, 1996, 44(5): 1280-1284.
[12] 康继韬, 俞雪钧, 谢东华, 等. 气相色谱/质谱测定水产品中氯霉素残留量[J]. 分析实验室, 2005, 24(7): 38-40.
[13] 谢孟峡, 刘媛, 邱月明, 等. 固相萃取-气相色谱质谱测定动物组织中氯霉素残留量[J]. 分析化学, 2005, 33(1): 1-4.
[14] NAGATA T, SAEKI M. Simultaneous determination of thiamphenicol, florfenicol, and chloramphenicol residues in muscles of animals and cultured fish by liquid chromatography[J]. Journal of Liquid Chromatography, 1992, 15(12): 2045-2056.
[15] 杨方, 余孔捷, 黄建生. 高效液相色谱法检测鳗鱼中氟苯尼考残留量[J]. 分析试验室, 2005, 24(2): 44-46.
[16] 占春瑞, 郭平, 陈振桂, 等. 超高效液相色谱法测定水产品中甲砜霉素和氟甲砜霉素残留[J]. 分析化学, 2008, 36(4): 525-528.
[17] 杨洪生, 孟勇, 张美琴, 等. 快速溶剂萃取-超高效液相色谱-串联质谱法同时测定水产品中氯霉素和氟苯尼考[J]. 理化检验: 化学分册, 2012, 48(11): 1353-1356.
[18] van de RIET J M, POTTER R A, CHRISTIE-FOUGERE M, et al. Simultaneous determination of residues of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in farmed aquatic species by liquid chromatography/mass spectrometry[J]. Journal of AOAC International, 2003, 86(3): 510-514.
[19] 彭涛, 李淑娟, 储晓刚, 等. 高效液相色谱/串联质谱法同时测定虾中氯霉素、甲砜霉素和氟甲砜霉素残留量[J]. 分析化学, 2005, 33(4): 463-466.
[20] 冯民, 魏云计, 朱臻怡, 等. 高相液相色谱-串联质谱法同时测定饲料中氯霉素、甲砜霉素与氟甲砜霉素残留量[J]. 分析测试学报, 2013, 32(1): 117-121.
[21] SHERIDAN R, POLICASTRO B, THOMAS S, et al. Analysis and occurrence of 14 sulfonamide antibacterials and chloramphenicol in honey by solid-phase extraction followed by LC/MS/MS analysis[J]. Journal of Agricultural and Food Chemistry, 2008, 56(10): 3509-3516.
[22] 刘艳琴, 王浩, 殷晓燕, 等. 高效液相色谱-电喷雾离子阱质谱测定乳品中氯霉素、甲砜霉素和氟甲砜霉素残留的研究[J]. 食品科学, 2008, 29(4): 344-346.
[23] 张凤清, 王岩松, 范世华, 等. 高效液相色谱-串联质谱法测定动物肌肉组织中氯霉素、甲砜霉素和氟苯尼考残留量[J]. 食品科学, 2010, 31(8): 248-251.
[24] 罗辉泰, 黄晓兰, 吴惠勤, 等. QuEChERS/液相色谱-串联质谱法同时测定鱼肉中30 种激素类及氯霉素类药物残留[J]. 分析测试学报, 2011, 30(12): 1329-1337.
[25] 万译文, 邓克国, 刘丽. 高效液相色谱-串联质谱法同时测定水产品中氯霉素、甲砜霉素和氟甲砜霉素的残留量[J]. 分析试验室, 2013, 32(5): 84-87.
[26] POSYNIAK A, ZMUDZKI J, NIEDZIELSKA J. Evaluation of sample preparation for control of chloramphenicol residues in porcine tissues by enzyme-linked immunosorbent assay and liquid chromatography[J]. Analytica Chimica Acta, 2003, 483(1/2): 307-311.
[27] 沈美芳, 赵文亚, 费志良, 等. 用酶联免疫法测定水产品中氯霉素的残留量[J]. 南京师范大学学报: 工程技术版, 2003, 3(2): 1-4.
[28] 庄宛, 叶玫. 动物性食品中氯霉素(CAP)残留检验方法概述[J]. 福建畜牧兽医, 2004, 26(1): 8-10.
[29] 戴军, 陈尚卫, 顾小红. HPLC测定牛奶和虾仁中氯霉素残留[J]. 无锡轻工大学学报, 2003, 22(6): 81-84.
[30] GIKAS E, KORMALI P, TSIPI D. Development of a rapid and sensitive SPE-LC-ESI MS/MS method for the determination of chloramphenicol in seafood[J]. Journal of Agricultural and Food Chemistry, 2004, 52(5): 1025-1030.
[31] MARIA D M A L, JOSE L M V, ROBERTO R G. Comparison of several extraction techniques for multiclass analysis of veterinary drugs in eggs using ultra-high pressure liquid chromatography-tandem mass spectrometry[J]. Analytica Chimica Acta, 2010, 661(2): 150-160.
[32] 李婷, 汤智, 洪武兴. 分散固相萃取-气相色谱法-质谱法测定含油脂肪食品中17 种邻苯二甲酸酯[J]. 分析化学, 2012, 40(3): 391-396.
[33] 张毅, 岳振峰, 蓝芳, 等. 分散固相萃取净化与液相色谱/串联质谱法测定牛奶中8 类禁用药物残留[J]. 分析化学, 2012, 40(5): 724-3729.
[34] 会恩健, 盛振华, 方文忠, 等. QuEChERS法联合在线GPC-GC-MS测定独活中27 种农药残留[J]. 中国实验方剂学杂志, 2013, 19(8): 138-141.
[35] 严忠雍, 张小军, 梅光明, 等. N-丙基乙二胺快速净化结合UPLCMS/MS测定水产品中氯霉素残留量[J]. 中国渔业质量与标准, 2013, 3(1): 65-69.
Determination of Chloramphenicol and Florfenicol in Fishery Products by Using Dispersive Solid Phase Extraction and Gas Chromatography
HU Hong-mei1, GUO Yuan-ming1,*, LEI Ke2, ZHANG Xiao-jun1, YAN Zhong-yong1, HE Yi-na1, YOU Ju-ju1, LIU Qin1
(1. Key Laboratory of Mariculture and Enhancement of Zhejiang Province, Marine Fishery Institute of Zhejiang Province, Zhoushan 316100, China; 2. School of Fishery, Zhejiang Ocean University, Zhoushan 316000, China)
An efficient, sensitive, and low matrix interference method for the determination of chloramphenicol (CAP) and florfenicol (FF) in fishery products using gas chromatography-electron capture detector (GC-ECD) has been developed. Samples were treated by extraction with ethyl acetate and n-hexane purification. Further cleanup of the extracts was processed by a modified dispersive solid phase extraction procedure. Then, the purif i ed solutions were derivatized with slilyl reagents. The linearity of the method ranged from 1.5 to 100 μg/L for CAP, and from 6 to 400 μg/L for FF, with correlation coeff i cients ranging between 0.998 1 and 0.999 7. The limits of detection were 0.1–0.3 μg/kg. The recoveries of spiked CAP and FF with external calibration method at different concentration levels in different sample matrixes (crucian, blue crab, and Penaeus vannawei) were 82%–106%, 87%–111%, and 91%–98%, respectively, with relative standard deviations of 2.3%–4.9%, 2.4%–4.5%, and 1.4%–4.1% (n = 5), respectively. Thus it is concluded that this method can be successfully applied for the determination of choramphenicols (CAPs) in fishery products.
dispersive solid phase extraction; gas chromatography-electron capture detector; fisher products; choramphenicols
S859.84
A
1002-6630(2014)08-0231-05
10.7506/spkx1002-6630-201408046
2013-07-31
浙江省科技计划项目(2012F30021;2012F30026);
浙江省“农产品安全标准与检测技术”重点科技创新团队项目(2010R50028);浙江省自然科学基金项目(LQ13C200004)作者简介:胡红美(1986—),女,工程师,硕士,研究方向为渔业环境与水产品质量安全。E-mail:huhm@zju.edu.cn
*通信作者:郭远明(1977—),男,高级工程师,硕士,研究方向为渔业环境与水产品质量安全。E-mail:yuanming_guo@126.com



